
FDA新药许可申请、审核及管理简介
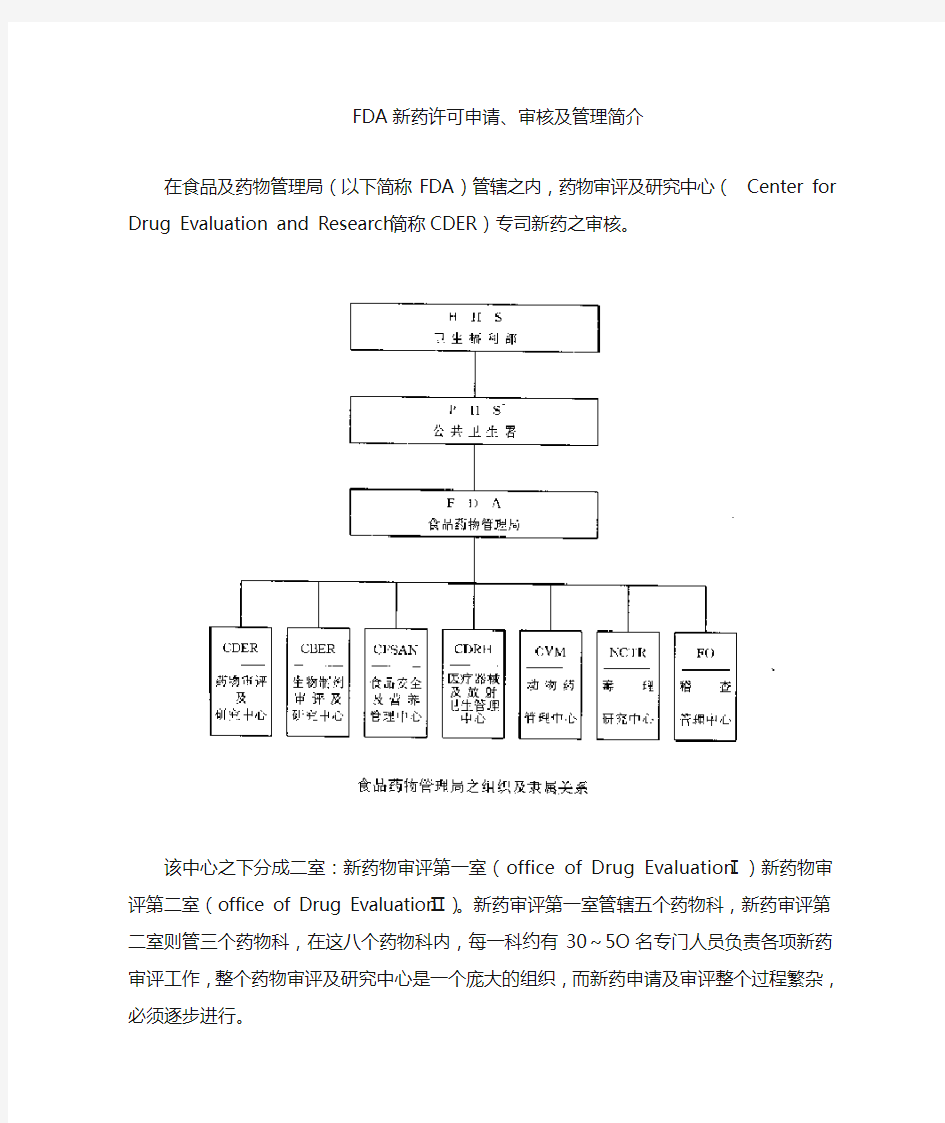
在食品及药物管理局(以下简称FDA)管辖之内,药物审评及研究中心(Center for Drug Evaluation and Research,简称CDER)专司新药之审核。
该中心之下分成二室:新药物审评第一室(office of Drug EvaluationⅠ)新药物审评第二室(office of Drug EvaluationⅡ)。新药审评第一室管辖五个药物科,新药审评第二室则管三个药物科,在这八个药物科内,每一科约有30~5O名专门人员负责各项新药审评工作,整个药物审评及研究中心是一个庞大的组织,而新药申请及审评整个过程繁杂,必须逐步进行。
药物审评及研究中心(Center for Drug Evalution and Research,CDER)之组织说明如下:
1.秘书室(Executive Secretariant Staff)
2.总务室(Office of Management)
(l)药物资记中心(Drug Information Resource)
(2)医学图书馆(Medical Library)
(3)总务及王计(Management and Budget)
(4)资讯系统设计(Information System Design)
3.专业进修室(Professional Development)
4.顾问团(Advisors and Consultants Staff)
5·前导性新药审评(Pilot Drug Evaluation)
6.非处方药审评室(office of OTC Drug Evaluation)
(1)单篇非处方药审评室(MonograPh Review Staff)
(2)非处方药政策性科(OTC Drug Policy Staff)
(3)医学审评科(Medical Review Staff)
7.非专利处方药室(office of Generic Drugs)
(1)化学第一科(ChemistryⅠ)
(2)化学第二科(ChemistryⅡ)
(3)生体相等性科(Bioequivalence)
8.研究发展室(Office of Research Resources)
(1)研究及试验科(Research and Testing)
(2)药品分析科(Drug Analysis)
(3)生体药学科(Biopharmaceutics)
(4)临床药理科(Clinical pharmacology)
9.新药标准室(Office of Drug Standards)
(l)新药市场广告及信息科(Drug Marketing,Advertising and Communications)10.新药合法性室(Office of Compliance)
(1)新药信息科(Drug Labeling Compliance)
(2)新药品质审核科(Drug Quality Evaluation)
(3)新药产品及制造品质科(Mahufacturing and Product Quality)
(4)科学性侦查科(Scientific Investigations)
(5)新药管理科(Regulatory Affairs)
11.新药流行学及统计生物学室(Office of Epidemiology and Biostatistics)
(1)流行学及监视科(Epidemiology and Surveillances)
(2)生物统计科(Biometrics)
12.新药审评第一室(Office of Drug EvaluationⅠ,ODEI)
(1)心脏药物科(Division of Cardio-Renal Drug Products)
(2)神经药物科(Division of Neuropharmacological Drug Products)
(3)肿瘤肺药物科(Division of Oncology and Pulmonary Drug Products)
(4)影像、手术及牙药物科(Division of Medical Imaging Surgical and Dental Drug Products)
(5)胃肠及凝血药物科(Division of Gastrointestinal and Coagulation Drug Products)
13.新药审评第二室(Office of Drug Evaluation Ⅱ, ODE Ⅱ)
(1)新陈代谢及内分泌药物科(Division of Metabolism and Endocrine Drug ProductS)
(2)抗传染药物科(Division of Anti-Infective Drug Products )
(3)抗病毒药物科(Division of Anti—Viral Drug ProductS)
二、新药申请
(一)药物的定义
依据联邦食品药物及化妆品法第二章第201节,药物的定义如下:
l.美国药典,同种治疗法药典,或者国家处方集(National Formulary)中所列的物质。
2.任何物质用于人或动物以利诊断、治愈、治疗、预防疾病。
3.任何物质用于改变人体或动物构造或功能。
4.任何物质属于上述1、2或3的一部分。
药物定义范围广泛,本文着重于人用新药,不包括生物制剂在内。
(二)新药申请手续
新药申请可分为两个阶段,即新药临床试验申请。(Investigational New Drug Application,简称IND)及新药上市许可申请(New Drug Application,简称NDA)。
1.新药临床试验申请(IND)
一般新药试验申请的目的是请求FDA核准进行第一次人体临床试验,不管是否已在美国之外其他各国进行人体试验,只要是在美国申请,需填Form FDA1571表。及Form FDA1572表。
新药试验申请所提出的内容应包括新药的物理化学性质、临床前(preclinical)或非临床(nonclinical),以及健康志愿者(healthy volunteer)或病人使用的经验[若是已在美国以外之国家取得人体试验结果,最重要的资料是第一次在美国进行人体试验的计划书(clinical protocol)]。进一步说明,新药试验申请至少应包括下列资料:
(1)药物的化学名称、分子式、构造、投药方式(口服、注射或其他方法),如无商标名称trade name)亦可先用公司暗码名称(code name)代替。
(2)主要成分及所知之非主要成分(没作用的成分)及所测得之杂物(impurities),并列出成分组成。
(3)供应药物公司的地址。
(4)药物来源,工厂设备(facilities)及质量控制(controls)的资料以说明如何制造、包装等,以确保药物的认定(identity)、效力(strength)、质量(quality)及纯度(purity)。
(5)所有临床前之动物药理实验、毒性试验资料、动物毒性试验,须遵照优良实验室规范(GLP)。若未遵照是项规定,则应加以说明。
(6)一份依据临床前动物实验的结果—确定该新药物第一次用于人体实验相当安全(reasonably safe)的声明。
(7)一份提供所有相关资料—包括其可能发生副作用之临床研究员手册(Clinical Investigator’s Brochure)。
(8)一份第一次在美国进行人体试验计划书(Protocol)—该计划书应阐明实验目的、实验手续、药物授予法、参加实验者合格条件及不合格条件(inclusion and exclusion criteria)、处理特别反应或过度反应之方式、实验室血液及尿液检查等.
(9)一份说明书(包括提供学历、经历及医学经验等文件)—证明医生及专业人员有足够能力执行人体试验。
(10)经过研究机构审评委员会(Institutional Review Board)的核准,进行人体试验计划之文件。
(11)一份同意于向FDA提出人体试验申请日起30天开始进行人体试验之声明书。
正如Form FDA1571规定,除非经FDA特准,一般而言,FDA收到新药试验申请日起30天内是不能进行人体试验的。新药试验申请送达相关的药物审评科评审后,如无问题,FDA也不会通知公司,亦即新药试验公司于30天期限未收到FDA通知有任何异议(objection)时此新药试验申请(IND)即开始生效(in effect)。换言之食品药物管理局并非“核准”(Approve)IND,而是“反对”而已。如FDA对该新药申请之临床试验持不同
意见,则有临床试验暂缓进行(Clinical hold)之判定,此时公司应充分合作,而补足FDA 所要求的资料或实验。
新药试验如无法确定向FDA那一科(Division)申请时,可先以电话查询。如申请之新药试验有两种药物,一般依具主要疗效(Primary mode。fuction)之药物来决定。如药物与生物制剂(biologics)或医疗器材(medical device)合并使用时,大致亦依主要疗效(Pri -mary mode fuction),或经过不同的中心(CDER,CBER或CDRH)讨论而决定管辖权,详细情况请参考,Federal Register,Docket No.91N—0257 Vol 56,No.225 November 21,1991。
《药品注册管理办法》(局令第28号) 培训复习题 一、填空题 1、《药品注册管理办法》(局令第28号)根据《中华人民共和国药品管理法》、《中华人民共和国行政许可法》、《中华人民共和国药品管理法实施条例》制定。 2、药品注册,是指国家食品药品监督管理局根据药品注册申请人的申请,依照法定程序,对拟上市销售药品的安全性、有效性、质量可控性等进行审查,并决定是否同意其申请的审批过程。 3、药物临床试验的受试例数应当符合临床试验的目的和相关统计学的要求,并且不得少于《药品注册管理办法》附件规定的最低临床试验病例数。 4、仿制药申请,是指生产国家食品药品监督管理局已批准上市的已有国家标准的药品的注册申请;但是生物制品按照新药申请的程序申报。 5、补充申请,是指新药申请、仿制药申请或者进口药品申请经批准后,改变、增加或者取消原批准事项或者内容的注册申请。 6、药品注册工作应当遵循公开、公平、公正的原则。 7、药物临床前研究应当执行有关管理规定,其中安全性评价研究必须执行《药物非临床研究质量管理规范》。 8、国家食品药品监督管理局应当执行国家制定的药品行业发展规划和产业政策,可以组织对药品的上市价值进行评估。 9、药品注册过程中,药品监督管理部门应当对非临床研究、临床试验进行现场核查、有因核查,以及批准上市前的生产现场检查,以确认申报资料的
真实性、准确性和完整性。 10、在药品注册过程中,药品监督管理部门认为涉及公共利益的重大许可事项,应当向社会公告,并举行听证。 11、申请人应当按照国家食品药品监督管理局规定的格式和要求、根据核准的内容印制说明书和标签。 12、两个以上单位共同作为申请人的,应当向其中药品生产企业所在地省、自治区、直辖市药品监督管理部门提出申请;申请人均为药品生产企业的,应当向申请生产制剂的药品生产企业所在地省、自治区、直辖市药品监督管理部门提出申请;申请人均不是药品生产企业的,应当向样品试制现场所在地省、自治区、直辖市药品监督管理部门提出申请。 13、药物的临床试验(包括生物等效性试验),必须经过国家食品药品监督管理局批准,且必须执行《药物临床试验质量管理规范》。药品监督管理部门应当对批准的临床试验进行监督检查。 14、申请新药注册,应当进行临床试验。临床试验分为I、II、III、IV期。 15、临床试验用药物应当在符合《药品生产质量管理规范》的车间制备。制备过程应当严格执行《药品生产质量管理规范》的要求。申请人对临床试验用药物的质量负责。 16、在新药审批期间,新药的注册分类和技术要求不因相同活性成份的制剂在国外获准上市而发生变化。 17、申请人完成药物临床试验后,应当填写《药品注册申请表》,向所在地省、自治区、直辖市药品监督管理部门报送申请生产的申报资料,并同时向中国药品生物制品检定所报送制备标准品的原材料及有关标准物质的研究资料。
药品质量安全管理规定 第一章总则 第一条总体目的 为保障消费者的用药安全,规范企业药品质量安全的管理,维护公司产品良好的质量形象,保证企业各项经营工作的正常顺利开展,特制定《药品质量安全管理规定》(以下简称“本规定”)。 第二条工作原则 公司统一领导、策划和监控,并提供资源支持;各部门重视并予以落实,做到组织有力,处理迅速得当,汇报及时,改进有效合理。 第三条适用范围 本规定适用*****有限公司。 第四条编制依据 《中华人民共和国药品管理法》、《药品不良反应报告和监测管理办法》、《药品召回管理办法》 第二章分类与概念 第五条药品质量安全管理包括: 1.药品生产运营过程安全评价管理 全方位进行检查生产运营是否符合法律法规的要求,是否具备合法完整的批准证明文件;评价生产工艺、质量控制等环节是否具备完善的质量安全保证体系,达到控制质量安全的目的。
2.药品质量安全日常事务及应急管理 对药品上市流通后可能产生的质量安全事务进行全方位的管理,包括: 1)不良反应监测管理 2)药品召回管理 3)质量问题和事故管理 4)药品监管信息管理 同时将严重不良反应、质量事故以及重大监管信息和其他有关药品的突发事件作为药品质量安全应急事务管理。 3.药品质量安全隐患预警管理 通过安全评价、现有的外部质量安全事务信息进行评估、分析,对可能造成质量安全风险的隐患通过提示、通报、警示等手段,提醒安全责任人采取必要的措施,消除或降低药品质量安全风险并及时跟踪反馈。 第三章组织管理 第六条领导小组 企业应成立以总经理为组长的药品质量安全领导小组,负责对药品质量安全的领导管理、战略决策以及突发事件的应急指挥、组织协调。 第七条职能管理机构 企业设立有公司有关领导和研发、销售、质量、生产等相关部门人员组成各职能管理小组和药品重大安全事件应 急管理机构。
药品注册管理办法 第一章总则 第一条为保证药品的安全有效和质量可控,规范药品注册行为,根据《中华人民共和国药品管理法》(以下简称《药品管理法》)、《中华人民共和国药品管理法实施条例》(以下简称《药品管理法实施条例》)制定本办法。 第二条在中华人民共和国境内从事药物研制、申请药物临床研究、申请生产和进口药品及进行与此相关的药品检验、监督和注册管理的单位或者个人,必须遵守本办法。 第三条国家药品监督管理局主管全国药品注册管理工作,负责对药物临床研究、药品生产和药品进口的审批。 省、自治区、直辖市药品监督管理局受国家药品监督管理局的委托,按照本办法的规定,负责对申报的药品注册资料的完整性、规范性和真实性进行审核。 第四条国家鼓励和支持研究创制新药。对创制的新药、治疗疑难危重疾病的新药给予快速审批。 第五条药品注册申请人和药物研究机构应具备完全民事行为能力。 进口药品注册须由国外制药厂商驻中国的办事机构或其在中国的注册代理办理。 药品注册申请人及代理药品注册的机构,须是在中国工商
行政管理部门注册登记的合法机构;代理药品注册的机构应具有相应的专业技术人员并取得药品注册申请人的授权。 办理药品注册申请事务的人员应熟悉药品注册管理法律法规及技术要求。 第二章药品注册的申请与受理 第六条药品注册申请分为新药申请、简略申请和补充申请。 新药申请系指未在我国上市销售过的药品的注册申请。已上市药品改变剂型、改变给药途径或制成新的复方制剂的,按新药管理。 简略申请系指已有国家标准的药品的注册申请。中药材、中药饮片、中药提取物的注册申请,按简略申请管理。 补充申请系指经国家药品监督管理局受理或批准新药申请、简略申请后,改变、增加或取消、注销原申请事项或内容的注册申请。 第七条新药注册申请应按照临床研究和生产(进口)两个阶段进行。 按简略申请注册的药品,一般可直接进入申请生产的阶段。如须进行临床研究的,应按照临床研究和生产(进口)两个阶段进行。 特殊情况下,对用于治疗严重危害人体健康疾病的药品,经国家药品监督管理局批准,药品注册申请人可直接申请生产(进口)注册。 第八条按新药申请办理注册的,药品注册申请人应向国家药品监督管理局委托的省、自治区、直辖市药品监督管理局提
FDA新药许可申请、审核及管理简介 在食品及药物管理局(以下简称FDA)管辖之内,药物审评及研究中心(Center for Drug Evaluation and Research,简称CDER)专司新药之审核。 该中心之下分成二室:新药物审评第一室(office of Drug EvaluationⅠ)新药物审评第二室(office of Drug EvaluationⅡ)。新药审评第一室管辖五个药物科,新药审评第二室则管三个药物科,在这八个药物科内,每一科约有30~5O名专门人员负责各项新药审评工作,整个药物审评及研究中心是一个庞大的组织,而新药申请及审评整个过程繁杂,必须逐步进行。
药物审评及研究中心(Center for Drug Evalution and Research,CDER)之组织说明如下: 1.秘书室(Executive Secretariant Staff) 2.总务室(Office of Management) (l)药物资记中心(Drug Information Resource) (2)医学图书馆(Medical Library) (3)总务及王计(Management and Budget) (4)资讯系统设计(Information System Design) 3.专业进修室(Professional Development) 4.顾问团(Advisors and Consultants Staff) 5·前导性新药审评(Pilot Drug Evaluation) 6.非处方药审评室(office of OTC Drug Evaluation) (1)单篇非处方药审评室(MonograPh Review Staff) (2)非处方药政策性科(OTC Drug Policy Staff) (3)医学审评科(Medical Review Staff) 7.非专利处方药室(office of Generic Drugs) (1)化学第一科(ChemistryⅠ) (2)化学第二科(ChemistryⅡ) (3)生体相等性科(Bioequivalence) 8.研究发展室(Office of Research Resources) (1)研究及试验科(Research and Testing)
药品安全工作总结 药品安全工作总结 在食品挂牌工作上,我局领导不等不靠,主动与县政府主要领导汇报有关情况,取得 了县领导的大力支持下,并拨付开办费2万元。x月x日上午,市局xxx局长与县政府 xxx县长为xx县食品药品监督管理局揭牌。在揭牌仪式上,市政府、市局、县四套班子主要领导应邀出席揭牌仪式,县政府主管副县长xxx同志代表县四大班子做了讲话。xx县食品药品监督管理局成立后,在继续行使原药品监督管理职能的基础上,增加了食品、化妆品、保健品综合监督、组织协调和依法组织开展对重大食品安全事故进行查处的新职能。 xx县食品药品监督管理局挂牌,是全市县级局第一家,同时也标志着全市县级食品药品监管工作进入一个新的发展阶段。 9月25日,灵宝市组织市食安办、食品药品监督管理局、工商局、粮食局、农业局等部门对我市6个行业、36个点的国庆节前食品安全进行联合执法检查,随机抽查了嘉佰利纯净水、珍桂坊面包店、鑫源果业、灵宝市实验二中、农垦奶牛场、永辉果业、粮丰公司 等企业。在生产加工单位了解市场供应、检查生产流程、产品质量及日常监管等情况,查 证照,看台账,在餐饮服务单位,对企业的执行食品索票索证和台帐登记等工作制度的情 况和食品采购、餐具消毒、烹饪加工、环境卫生进行了认真细致地重点检查。对检查中发 现的问题当即提出整改意见,要求经营者限期整改。确保国庆节期间食品安全。 按照省、市局食品药品工作会议精神和XX县委、政府对食品安全工作的要求,在机 构改革未到位之前,继续履行好食品安全综合职责,进一步完善食品安全机制,积极协调 相关部门加强食品安全,深入开展食品安全专项整治,有效保障了全县人民群众饮食安全。 今年我局在区政府的统一安排下,与区卫生、质检、农委等部门在区政府门前,举行 了大型广场宣传咨询活动,以横幅、展板、发放宣传材料、现场咨询和实物比较等形式, 向过往群众宣传食品安全法律法规、食品安全专项整治成果以及食品安全科普知识,共发 放宣传材料*****多份,咨询服务群众****多人次,收到了良好的宣传效果。 为维护食品市场秩序,保障灵宝75万群众生命财产安全,切实做到中秋、国庆期间 食品安全监管工作不懈怠,我市开展了以下工作: 继续做好食品安全工作。一是配合有关部门进一步加强食品安全监管力度,帮助企业 建立食品安全信用体系长效机制。二是完善食品安全信用体系行业评价制度。三是继续帮 助和指导企业开展HACCP及其质量体系的认证,力争2020年认证企业达到80%。四是协助有关部门开展绵阳市食品安全生产示范单位建设活动。五是继续做好食品安全宣传月活动。
药品注册管理办法 (征求意见稿) 目录 第一章总则 第二章基本制度和要求 第三章药品上市注册 第一节药物临床试验 第二节药品上市许可 第三节关联审评审批 第四节药品注册核查 第五节药品注册检验 第四章药品加快上市注册 第一节突破性治疗药物程序 第二节附条件批准程序 第三节优先审评审批程序 第四节特别审批程序 第五章药品上市后变更和再注册 第一节药品上市后研究和变更 第二节药品再注册 第六章受理、补充资料和撤审 第七章争议解决 第八章工作时限 第九章监督管理 第十章法律责任 第十一章附则 —1 —
第一章总则 第一条【法律依据】为规范药品注册行为,保证药品的安全、有效和质量可控,根据《中华人民共和国药品管理法》(以下简称《药品管理法》)《中华人民共和国中医药法》《中华人民共和国疫苗管理法》(以下简称《疫苗管理法》)《中华人民共和国行政许可法》《中华人民共和国药品管理法实施条例》,制定本办法。 第二条【适用范围】在中华人民共和国境内以药品上市为目的,从事药品研制、注册及其监督管理活动,适用本办法。 第三条【药品注册定义】药品注册,是指药品注册申请人(以下简称申请人)依照法定程序和相关要求提出药品注册事项,药品监督管理部门基于法律法规和现有科学认知进行安全性、有效性和质量可控性等审查,作出是否同意其药品注册事项及其管理的过程。 申请人取得药品注册证书后,为药品上市许可持有人(以下简称持有人)。 第四条【药品注册事项】药品注册包括药物临床试验申请、药品上市许可申请、补充申请、再注册申请等许可事项,以及其他备案或者报告事项。 第五条【药品注册申请类别】药品注册申请类别,按照中药、化学药和生物制品等进行分类。 中药注册分类包括中药创新药、中药改良型新药、古代经典名方中药复方制剂、同名同方药等。 化学药注册分类包括化学药创新药、化学药改良型新药、仿制药等。
2017年药剂科质量与安全管理工作计划 一、药事管理 进一步完善制定医院基本用药目录。根据我院新的基本药物目录,由我院药事管理与药物治疗学委员会进行药品遴选,制定出我院2015年的基本用药目录,并保证目录内的药品供应,保证临床的用药需求。 二、药品管理 加强药品质量管理,保障患者用药安全,由于药剂科承当着全院的药品供给工作,是医院的重要的临床辅助科室。严格按照我院与医药公司签订的购销协议、招标采购制度采购药品,严把药品质量关,确保购进药品的质量,及时掌握临床科室用药需求,确保临床用药的供应。药品质量与安全管理小组进一步规范药品的购进、验收、在库养护等环节,管理小组成员每个月对科内工作流程及各岗位的工作质量进行抽查,并催促科室工作人员认真执行各项质量管理制度,从而有效保证了我院药品质量,保障了患者的用药安全;加强麻醉药品、精神药品的管理工作,严格执行麻醉药品、一类精神药品的“五专”管理;每月对各病区、手术室、各诊室的麻醉药品、一类精神药品、高危药品、备用基数药品、抢救药品的管理使用情况组织检查,对存在的问题提出改进意见,并督促整改;每季度召开质量与安全管理会议,对本部门质量与安全管理进行检讨,对全院药学质量与安全进行总结分析;提高药房窗口服务,宣传用药知识,让患者充分感受药师的责任心。 三、积极展开药品不良反应的监测。 在平常工作中,主动到临床搜集药品使用后的信息反馈。发现药品发生不良反应时,协助临床做好药品不良反应的处理工作并查找缘由,如与药品质量有关的,及时更换厂家,以保证临床用药安全。依照药品不良反应的监测“可疑必报”的原则,我科及时做好药品不良反应/事件的互联网直报工作。
新版药品注册管理办法(2016-7-25修订稿)的解读 版本概况:发布日期:2016年7月25日,征求意见至2016年8月26日 药审机构的职能 原文:第十条:药审机构可根据产品风险和技术审评需要提出现场检查、样品注册检验等要求,并综合现场检查、样品注册检验等报告做出技术审评结论。 评析:这条看起来很平常。可是FDA就是这么干的。也就是说,民间猜测的CFDA 的模式会趋向于美国FDA的模式是真的。CDE可能变成一个大中心,独立进行受理,收回省局的受理、批准权。省局将只有日常监管和补充申请审核的权利。而且 CDE可以发起有因核查、复核检验。FDA并非以复核检验为申报必须条件,一切都是因风险而选择的。那以后中检院的职能会不会有所改变呢?这里的药审机构定义是CDE,注意,是CDE,因为规定了国家局的职能是建立科学规范、完善高效的审评审批体系,省局没这职能,所以不包含省局技术审评部门。 由此省局职能只剩下日常管理,上市监查,被“抄报”。上市许可,这次的征求意见稿里不分新药和还是仿制药,一句话:向食品药品监管总局提出上市申请,食品药品监管总局作出行政许可决定的过程。同时看一眼第一百零五条食品药品监管总局对药品有效性、安全性和质量可控性等没有影响的变更,实施备案管理。备案管理都是总局的了。省局注册处已名存实亡…… 药品注册申请类别 原文:第三条药品注册申请包括药物临床试验申请、药品上市申请、药品上市后注册事项变更的补充申请以及延续申请。 评析:药品注册申请类别已简化为临床申请、上市申请和补充申请三类,不再区分药品注册申请为新药还是仿制药 申请人主体资格 原文:第四条申请人是指提出药品注册申请并能依法承担民事责任的境内主体或者境外合法制药厂商。
仿制药的简略新药申请程序(ANDA Process)?前言 ?简略新药申请的指南文件 ?法律,法规,政策和程序 o美国联邦法典 o政策和程序手册 ?ANDA 表格和电子申请 ?药品开发和评审定义 ?药品开发方面的常见问题 ?相关主题 前言 An Abbreviated New Drug Application (ANDA) contains data which when submitted to FDA's Center for Drug Evaluation and Research, Office of Generic Drugs, provides for the review and ultimate approval of a generic drug product. 简略新药申请(ANDA)所包括的资料被交至FDA药品评审和研究中心所属的仿制药办公室,用于仿制药的评审和最终批准。Once approved, an applicant may manufacture and market the generic drug product to provide a safe, effective, low cost alternative to the American public.一旦批准,则申请者可以生产和销售该仿制药以向美国公众提供安全,有效,廉价的替代品。 A generic drug product is one that is comparable to an innovator drug product in dosage form, strength, route of administration, quality, performance characteristics and intended use. 仿制药在剂型,剂量,服用方式,质量,性能特征和用途方法都是和原创药物相当的。All approved products, both innovator and generic, are listed in FDA's Approved Drug Products with Therapeutic Equivalence Evaluations (Orange Book).所有被批准的药品,包括原创药和仿制药都列在FDA的具有相当疗效评估的已批准药品(橙皮书)上。Generic drug applications are termed "abbreviated" because they are generally not required to include preclinical (animal) and clinical (human) data to establish safety and effectiveness. 仿制药申请被称为“简略申请”,是因为它们基本上不需要临床前资料(动物实验)和临床资料(人体实验)来建立安全性的有效性。Instead, generic applicants must scientifically demonstrate that their product is bioequivalent (i.e., performs in the same manner as the innovator drug). 仿制药申请者必须要科学地论述他们的产品是生物等效的(也就是,和原创药同样的性能表现)One way scientists demonstrate bioequivalence is to measure the time it takes the generic drug to reach the bloodstream in 24 to 36 healthy, volunteers. 科学家论证生物等效性的一种方法就是测定仿制药到达24 ̄36位健康志愿者血流中的时间。This gives them the rate of absorption, or bioavailability, of the generic drug, which they can then compare to that of the innovator drug. 这个实验给出
安全用药相关管理制度 安全用药是一个动态过程,包括药品的采购、储存、保管、调配、使用等一系列环节。为加强药品管理,坚持以病人为中心的医院管理年服务理念,促进临床合理用药,避免药源性疾病的发生,最大限度地保障人民群众用药安全,根据《药品管理法》、《医疗机构药事管理暂行规定》等有关法律、法规,特制订我院安全用药管理制度。 1.科学、规范我院药事管理工作。药事工作是医疗工作的重要组成部分。要将医、药、护三个方面有机的结合起来,建立医师药师护士、药库药房病区的立方体空间体系,建立药剂科与医务科、护理部等管理部门的沟通机制,制定包括药品采购、储存保管、调配及使用等每一个环节的相关管理制度。全面保证用药安全。 2.执行《药品采购管理制度》和《新药采用管理制度》,执行新药遴选程序,坚持质量第一,按需进货,择优采购的原则,确保药品质量和购进的合法性。 3.执行《药品储存、保管管理制度》、《效期药品管理制度》、《高危药品管理制度》、《相似药品管理制度》等,遵守药品储存管理规范,执行各项工作程序,按照药品性能,对药品实行分区、分类管理。对高危药品做好醒目标示,避免差
错事故的发生。根据药品储存条件的要求,将药品分别存放于常温环境(0~30℃)、阴凉环境(≦20℃)、冷柜(2~10℃)中,并保持相对温度达标。 4.严格执行《麻醉药品、精神药品管理制度》,按照有关规定购进麻醉药品、第一类精神药品,保持合理库存。入库验收必须货到即验,至少双人开箱,清点验收到最小包装,验收记录双人签字。药库、药房要做到五专。 5.医师要严格按照《处方管理办法》的要求,根据医疗、预防、保健需要,按照诊疗规范、药品说明书中的药品适应症、药理作用、用法、用量、禁忌、不良反应和请注意事项等开具处方。并使用级药品通用名称和复方制剂药品名称。按照卫生部制定的麻醉药品和精神药品临床应用指导原则,开具麻醉药品、第一类精神药品处方。 6.医师必须尊重患者对应用药物进行预防、诊断和治疗的知情权。医务人员发现可能与用药有关的严重不良反应,在做好观察记录的同时,必须按规定及时上报。 7.药师必须严格执行各项操作规程和《处方管理制度》,认真审查和核对,必须做到四查十对,确保发现药品的准确、无误。并进行安全用药指导。
药剂科药品质量与安全管理 药剂科药品质量管理工作包括药品质量与安全管理和药学工作质量与安全管理,其管理内容主要是指对药品采供、药品调剂、药品存储等工作的全过程进行质量与安全管理(微观上的管理)和对药学工作的各部门、各环节进行全面质量与安全管理(宏观上的管理)。 药剂科药品质量与安全管理方案如下: 一、药剂科药品质量与安全管理工作制度 (一)按照《中华人民共和国药品监督管理法》的要求,进一步规范药剂科药房药库。 (二)严格按照我院的药品招标采购制度采购药品,严把药品质量关。 (三)加强专业人员的药学知识和技能的培训和考核,以便能更好地开展相关业务和完成我院布置的工作任务。 (四)认真执行《处方管理办法》,调配处方时能够做“四查十对”,并且复核双签字,确保处方合格率大于98%,不调配错一张处方不发错一颗药。 (五)做好处方点评工作,定期抽查处方和病历,对不合理用药甚至错误用药或滥用药的行为及时反馈到当事医生,严重的汇报到我院相关部门,进一步规范我院的合理用药。 (六)加强医德医风教育,树立全心全意为人民服务的崇高思想,以人为本,给患者提供优质药学服务。 (七)按照二级甲等医院的质量标准,完成各项工作任务。 (八)坚决执行我院的文件精神和高标准高质量地完成我院布置的各项工作任务。
二、药剂科药品质量与安全管理组织及任务 1.药品质量与安全管理小组的组成: 在院药事管理与药物治疗学委员会的领导下,药剂科成立药品质量与安全控制小组(简称质控小组)。组长由药剂科主任XXX担任,副主任XXX担任副组长。各药房负责人及临床药师任组员。 组长:XX 副组长:XXX 组员:XXX 2.质控小组的主要任务 (1)定期(每月)检查、考核全科药品质量、工作质量和管理情况,及时分析、处理存在的问题,督促全科质量与安全标准的落实。 (2)定期(每月)检查调剂室和药库毒、麻、精神、贵重等特殊药品管理情况,有无“四无”药品,有无假、劣、过期失效和变质药品。 (3)定期(每月)检查护士工作站药品质量和特殊药品管理情况(主要由临床药师执行,质控小组每季度检查一次,督导持续性改进情况)。 (4)定期(每月)到临床各科室了解医护人员及病人对药剂工作意见,介绍新药,收集有关不良反应的情况,不断提高药剂工作质量,确保临床用药安全有效。(5)定期召开质量与安全管理会议,对本部门的质量与安全管理进行检讨,对全院的药学质量与安全进行总结分析,每年度至少一次。 (6)定期向临床科室通报医院临床用药安全监测结果,提出整改建议。对从事质量和安全管理的员工有质量管理基本知识和基本技能培训教育。 三、药学工作质量与安全管理考核指标(质控指标) 根据《药品管理法》、《医疗机构药事管理办法》和《医院工作质量管理考核》等有关文件的规定和要求,结合本科工作实际,制定以下质量与安全管理考核指标: 1.调剂工作:各项工作均符合要求 (1)门诊处方总数复核率100%。
新药品注册管理办法规定修订稿
附件 药品注册管理办法(修订稿) 第一章总则 第一条为保证药品的安全、有效和质量可控,规范药品注册行为,根据《中华人民共和国药品管理法》(以下简称《药品管理法》)、《中华人民共和国行政许可法》(以下简称《行政许可法》)、《中华人民共和国药品管理法实施条例》(以下简称《药品管理法实施条例》),制定本办法。 第二条在中华人民共和国境内从事药品注册,及其涉及的药物研制和监督管理,适用本办法。 第三条药品注册,是指药品注册申请人(以下简称申请人)依照法定程序和相关要求提出申请,药品监督管理部门对拟上市药品的安全性、有效性、质量可控性等进行审查,作出行政许可决定的过程。 药品注册申请包括药物临床试验申请,药品上市许可申请、上市后补充申请及再注册申请。 第四条申请人,是指提出药品注册申请并承担相应法律责任的机构。 境内申请人应当是在中国境内合法登记并能独立承担法律责任的药品生产企业或研发机构。 境外申请人应当是境外合法制药厂商,应当指定境内具备相应质量管理、风险防控、责任赔偿能力的法人办理注册事项。 第五条国家药品监督管理部门负责管理全国药品注册工作;统一受理和审查药品注册申请,依法进行许可。省、自治区、直辖市药品监督管理部门负责本行政区域内药品注册相关的监督管理工作。 第六条国家实行药品上市许可持有人制度。药品上市许可持有人对上市药品的安全性、有效性和质量可控性进行持续考察研究,履行药品的全生命周期管理,并承担法律责任。 第七条药品注册工作应当遵循公开、公平、公正的原则,实行相关人员公示制和回避制、责任追究制,受理、检验、审评、审批、送达等环节接受社会监督。 第八条国家药品监督管理部门药品审评机构(以下简称药品审评机构)建立临床主导的团队审评制度、项目管理人制度、与申请人会议沟通制度、专家咨询委员会公开论证重大分歧制度、审评结论和依据公开制度等。
第一章总则 第一条为进一步规范药品管理工作,明确各部门职责和工作流程,切实满足各部门的药品使用需求,特制订本制度。 第二条药品指用于预防、治疗、诊断人的疾病有目的地调节人的生理机能并规定有适应症或者功能主治、用法和用量的物质。本制度中所称药品分为两类 (一)普通药品:指中成药、化学原料药及其制剂, (二)特殊药品指麻醉药品、精神药品、医疗用毒性药品和放射性药品 第三条制度适用于下属检测中心的药品管理。 第二章药品管理相关部门及职责 第四条采购供应部是药品的采购统筹部门,负责统一管理各区域检测中心的药品采购、下单、验收工作,采购管理岗负责具体实施。“ 第五条设备物资部是药品的库存与盘点统筹部门.负责监督、指导各区域检测中心药品的库存与盘点 第六条区域检测中心内设的各检查科室是药品的需求和使用部门。由各检查科室负责人负责,主要职责为审核药品需求、负责药品的合理、合规使用 第七条区域检测中心核医学科是放射性药品的需求、储存和使用机构,由核医学科主任负责.主要职责为:放射性药品的下单、验收、储存管理工作 第八条区域检测中心综合管理部是药品的综合管理部门后勒保障岗具体实施主要职责为汇总各科室药品需求。提请签报、下单,参与验收、盘点、报损工作 第九条区域检测中心护士团队是普通药品、麻醉药品精神药品、医疗用毒性药品的储存和领用部门,由护士长负责。主要职责为负责普通药品、麻醉药品、精神药品、医疗用毒性药品的保管,保证储存质量开辰入库验收、盘点工作负责收集和评估各科室的领用需求,确保合规领用做好领用记录。 第十条区域检测中心财务团队是药品下单、盘点、报损管理工作的支持部门。
第三章药品的下单管理 第十一条区域检测中心根据年度预算和实际使用需求,在采购供应部签订的框架协议内进行下单操作。下单分为两种类型 (一)常规下单。每月由区域检测中心后勤保障岗汇总下一月度药品的需求 (二)特殊药品下单。其中普通药品、麻醉药品、精神药品、医疗用毒性药品由护士长负责;放射性药品由核医学科主任负责。 第四章药品的验收、入库管理 第十二条库房设立待验区、合格区、不合格区,药品应按区存放。药品到货后应先进入待验区,并根据验收结果进入合格区或不合格区,不合格药品严禁入库、上架。 第十三条区域检测中心收到药品后,应立刻组织验收工作: (一)普通药品、麻醉药品、精神药品、医疗用毒性药品由后勤保障岗、护士长双人当场验收; (二)放射性药品由后勤保障岗、核医学科主任双人当场验收。 验收人员应对照订单,核对药品名称、规格、单位、数量、批号、生产厂家等信息并填写验收单(附件3)由区域检测中心后勤保障岗提起EOA签报(附件2)。 第十四条到货药品应在有效期内。其中,有效期3年以上的,到货时药品的剩余有效期不得少于21个月;有效期为15年至3年(含)的,到货时药品的剩余有效期不得少于16个月,有效期为1年至1.5年(含)的,到货时药品的剩余有效期不得少于8个月。 第十五条验收过程中,如发现以下问题严禁入库 (一)药品包装内有异常响动和液体渗漏 (二)外包装出现破损、封口不牢、封条严重损坏等现象: (三)包装标识模糊不清或脱落 第五章药品的储存、领用管理 第十六条分类管理:在库药品应技性质、剂型分大类,再技药理作用系统分类存放。
新药研发流程 药物从最初的实验室研究到最终摆放到药柜销售平均需要花费12年的时间。进行临床前试验的5000种化合物中只有5种能进入到后续的临床试验,而仅其中的1种化合物可以得到最终的上市批准。 总的来说新药的研发分为两个阶段:研究和开发。这两个阶段是相继发生有互相联系的。区分两个阶段的标志是候选药物的确定,即在确定候选药物之前为研究阶段,确定之后的工作为开发阶段。所谓候选药物是指拟进行系统的临床前试验并进入临床研究的活性化合物。 研究阶段包括四个重要环节,即靶标的确定,模型的建立,先导化合物的发现,先导化合物的优化。 一、靶标的确立 确定治疗的疾病目标和作用的环节和靶标,是创制新药的出发点,也是以后施行的各种操作的依据。药物的靶标包括酶、受体、离子通道等。作用于不同的靶标的药物在全部药物中所占的比重是不同的。以2000年为例,在全世界药物的销售总额中,酶抑制剂占32.4%,转运蛋白抑制剂占16.0%,受体激动剂占9.1%,受体拮抗剂占10.7%,作用于离子通道的药物占9.1%等等。目前,较为新兴的确认靶标的技术主要有两个。一是利用基因重组技术建立转基因动物模型或进行基因敲除以验证与特定代谢途径相关或表型的靶标。这种技术的缺陷在于,不能完全消除由敲除所带来的其他效应(例如因代偿机制的启动而导致的表型的改变等)。二是利用反义寡核苷酸技术通过抑制特定的信使RNA对蛋白质的翻译来确认新的靶标。例如嵌入小核核糖核酸(snRNA)控制基因的表达,对确证靶标有重要作用。 二、模型的确立 靶标选定以后,要建立生物学模型,以筛选和评价化合物的活性。通常要制订出筛选标准,如果化合物符合这些标准,则研究项目继续进行;若未能满足标准,则应尽早结束研究。一般试验模型标准大致上有:化合物体外实验的活性强度;动物模型是否能反映人体相应的疾病状态;药物的剂量(浓度)——效应关系,等等。可定量重复的体外模型是评价化合物活性的前提。近几年来,为了规避药物开发的后期风险,一般同时进行药物的药代动力模型评价(ADME评价)、药物稳定性试验等。 三、先导化合物的发现 新药研制的第三步是先导化合物的发现。所谓先导化合物(leading compound),也称新化学实体(new chemical entity,NCE),是指通过各种途径和方法得到的具有某种生物活性或药理活性的化合物。因为目前的知识还不足以渊博到以足够的受体机制指导药物设计以使药物的合成不必使用预先已知的模型,所以,先导化合物的发现,一方面有赖于以上两步所确定的受体和模型,另一方面也成为了整个药物研发的关键步骤。一般来说,先导化合物主要有如下几个来源:对天然活性物质的挖掘、现有药物不良作用的改进以及药物合成心中间体的筛选等。目前,主要有两个获得新的先导化合物的途径。一是广泛筛选,这种毫无依据的方法在实际操作上其实是比较有效的。过去半个多世纪以来,由于这个原因,先导化合物的发现随机性很强,如从煤焦油中分离出的本份被发现具有抗菌作用因而被开发成为一系列诸如萨罗的抗生素;又如对染料中间体的筛选发现了苯胺以及乙酰苯胺具有解热镇痛作用,经改造得到了非那西丁和乙酰氨基酚等。近二十年来,计算机预筛被用于这一过程,大大加快了研究进程。
药剂科质量与安全管理2014年工作计划 一、进一步完善制定医院基本用药目录。根据我院新的基本药物目录,由我院药事委员会进行药品遴选,制定出我院2014年的用药基本目录,并保证目录内的药品供应,保证临床的用药需求,力争使基药使用占比达到35%。 二、认真执行药事管理相关制度,积极配合医院网络建设,建立完善的药品管理信息系统,完善药品查询系统,方便医护人员查询、获取正确的药品信息。 三、加强理论学习,提高全体人员的政治思想觉悟和业务素质。定期组织全科人员认真学习上级及院内各种文件精神,定期开展业务学习及服务技能的培训,并贯彻执行到位,自觉抵制行业不正之风,以提高窗口服务为己任,以质量第一,病人第一为理念,全心全意为人民服务。 四、加强抗菌药物管理,继续贯彻执行抗菌药物临床应用的有关规定,把抗菌药物各项指标力争控制在范围内,加强ⅰ类切口手术合理使用抗菌药物的管理。 五、加强药品管理,保证临床用药需求和患者用药安全。每月定期对科内工作流程及各岗位的工作质量进行抽查,并督促科室工作人员认真执行各项管理制度。加强药品质量管理,在购进验收、入库养护等环节严把质量关,杜绝假冒伪劣药品混入我院,避免因药品过期造成重大医疗事故和经济损失。每月对药库、药房、病区的药品储备质量、效期等进行检查,将检查结果汇总,发现问题及时妥善处理。 六、完善工作流程,防止发生差错事故。药房窗口服务工作是医院服务工作的重要组成部分,不仅是反映医院精神面貌和文明素质的窗口,更担负着保障人民群众用药安全的重大责任。本着对病人负责、对自己负责的态度,一定要完善工作流程,审方、发药由专人负责,调剂由专人负责,经过两道把关,基本上可以做到防止发生差错事故,从而减少病人的投诉,杜绝医疗事故的发生。 七、加强麻醉药品的管理和使用,组织有关人员认真学习“安徽省医疗机构麻醉药品和精神药品管理规定”。 1、建立麻醉药品和精神药品的采购、验收、贮存、保管、发放、调剂、使用、督查、报残损、销毁、丢失及被盗案件报告、值班、巡查等制度; 2、每季度对麻醉药品和精神药品管理进行检查,并有记录,及时纠正存在的问题、消除隐患;定期召开会议,对麻醉药品和精神药品的管理工作各环节存在的问题进行总结,提出改进意见,适时修订相关制度; 3、定期对涉及麻醉药品和精神药品使用和管理的医学、药学、护理、医疗行政管理、保卫等部门人员进行有关法律法规、部门规章、专业知识、职业道德等的培训和考核。 八、按照“处方点评制度”,每月进行一次处方点评,主要检查抗菌药的规范使用、用药的适宜性、药品的用量、处方的规范书写,重点对用药不适宜处方及大处方进行合理性分析评价,并在医院内部通报处方评价结果。 九、严格执行“双十制度”,每月统计销售排名前十位的药品,对连续销售排名前十位的药品,经院药事委研究同意后,分别给予降低购进价格和限制销售数量的处罚。 十、继续做好药品不良反应()及不良事件监测工作。充分发挥护士长的作用,提高医疗器械不良事件的上报工作,使此项工作有较大的进步。篇二:药剂科质量与安全管理制度药剂科质量与安全管理 1.药品质量与安全管理制度 2.药剂科药品质量与安全管理组 3.药剂科药品质量与安全管理组工作职责 4.药剂科药品质量与安全监控小组成员 5.药剂科药品质量与安全监控小组工作职责 药品质量与安全管理制度
郎溪县医疗机构药品安全管理操作办法为规范医疗机构药品使用管理,提高医疗机构的药品质量管理水平,确保医疗机构药品质量,保障人民群众用药安全有效,根据《中华人民共和国药品管理法》、《中华人民共和国药品管理法实施条例》、《药品不良反应报告和监测管理办法》《医疗机构药品监督管理办法》、《药品流通监督管理办法》、《安徽省药品和医疗器械使用监督管理办法》等,特制定郎溪县医疗机构药品安全管理操作办法。 一、机构与人员要求 (一)药品使用单位要设臵相应的药品管理机构或者确定专职药品质量管理人员,具体负责药品质量的管理工作。 (二)药品质量管理人员应具有药学技术职称或经县级食品药品监督管理部门专门培训合格。 (三)从事药品采购、验收、养护工作的人员应具有药士以上药学专业技术职称称或执业药师资格。 (四)直接接触药品的人员,应当每年进行健康检查并建立健康档案。患有精神病、传染病及其他可能污染药品疾病的人员不得从事直接接触药品的工作。 二、制度和职责要求
(一)制定与药品使用质量管理有关的文件,包括质量管理制度、职责、工作程序和相关记录。 (二)质量管理制度应包括以下内容: 1、各级药品质量管理岗位职责; 2、药品购进、验收、储存、养护、出库等环节的管理; 3、药品效期的管理;4、特殊药品的管理;5、不合格药品的管理;6、药品使用差错和事故报告与处理的管理;7、拆零药品的管理;8、与药品储存质量有关设施设备的管理;9、质量管理文件的管理;10、环境卫生和人员健康的管理;11、人员培训和考核的管理。 (三)质量职责应包括以下内容: 1、质量管理、购进、仓储、药品调剂人员职责; 2、药品质量管理负责人及质量管理、购进、仓储、药品调剂人员职责; 3、质量管理、购进、收货、验收、仓储、养护、药品调剂岗位职责。 (四)工作程序应包括以下内容: 1、质量管理文件管理的程序; 2、药品购进、收货、验收、仓储、养护、药品调剂等环节的程序; 3、药品出库退回的程序; 4、药品退出的程序; 5、不合格药品的确认及处理程序; 6、中药材、中药饮片养护的程序。 (五)药品使用质量管理工作记录必须清楚、完整、准确。具体包括:
XX镇食品药品安全管理工作制度为加强对全镇食品药品安全管理的综合监管和组织协调,建立协调、高效的食品药品安全管理协调机制,特制定XX镇食品药品安全管理工作制度。 一、会议制度 每半年召开一次食品药品安全管理站工作会议,传达上级有关食品药品安全的法律法规和方针、政策、指示精神,统筹协调本镇食品药品安全监管工作,如遇特殊情况可随时召开。 二、投诉举报和处理制度 (一)镇食品药品安全管理站及有关部门要分别设立并向社会公开食品药品安全案件举报电话,安排专人负责受理投诉举报工作。 (二)按照首接负责的原则,对每起投诉举报要认真记录、及时处理或报县食品安全委员会办公室,做到件件有回音,事事有着落。 三、信息收集、发布及报告制度 为第一时间掌握全镇食品药品安全信息,确保信息反映灵敏、准确、及时、有效,建立XX镇食品药品安全管理站信息报告联络制度。管理站负责收集、汇总和分析整理食品药品安全信息,每月向县食品药品监督管理局报送,并实行零报告制度。 (一)食品药品安全信息网络组织:食品药品安全管理站要确定一名联络员,负责食品药品安全信息联络工作。建立信息审核报送制度,由管理站负责人审核签字后方可上报。建立食品药品安全信息网络,随时联络与定期召开会议相结合。
(二)食品药品安全信息报告内容:主要包括食品药品安全的监管执法情况、存在问题及拟采取的措施;专项整治进展情况;食品药品安全动态;重大事故查处信息等。各部门报送的信息要经过加工、整理、汇总,并简要评析分析本系统和本部门工作情况。 (三)安全信息报告时间:工作信息、技术信息每季度报告一次;食品药品安全动态信息随时报告。 (四)遇有重大食品药品安全案件或突发事件,要及时向县食品药品监督管理局报告。接报后由管理站组织协调处理并按照规定程序上报。 四、宣传培训制度 组织有关单位按照各自职责分工,开展食品药品安全宣传活动,采取多种形式,动员社会参与,营造良好的舆论氛围。根据实际工作需要,每年开展食品药品安全协管员、信息员培训工作,不断提高食品药品安全协管员、信息员业务素质和工作水平。 五、档案管理制度 做到镇管理台帐档案化,各类文件资料归口管理,档案资料盒子化管理,要分别有县委、县政府文件盒、县食品安全委员会及其办公室文件盒、镇党委政府文件盒、目标责任书资料盒、食品安全法律法规盒、专项整治资料盒、食品安全工作报表盒及各行政村资料盒。 六、督察督办和责任追究制度 (一)定期对所部署工作的落实情况进行监督检查,重点对食品放心工程进行监督检查。对措施不力,达不到规定要求的进行督察督办。