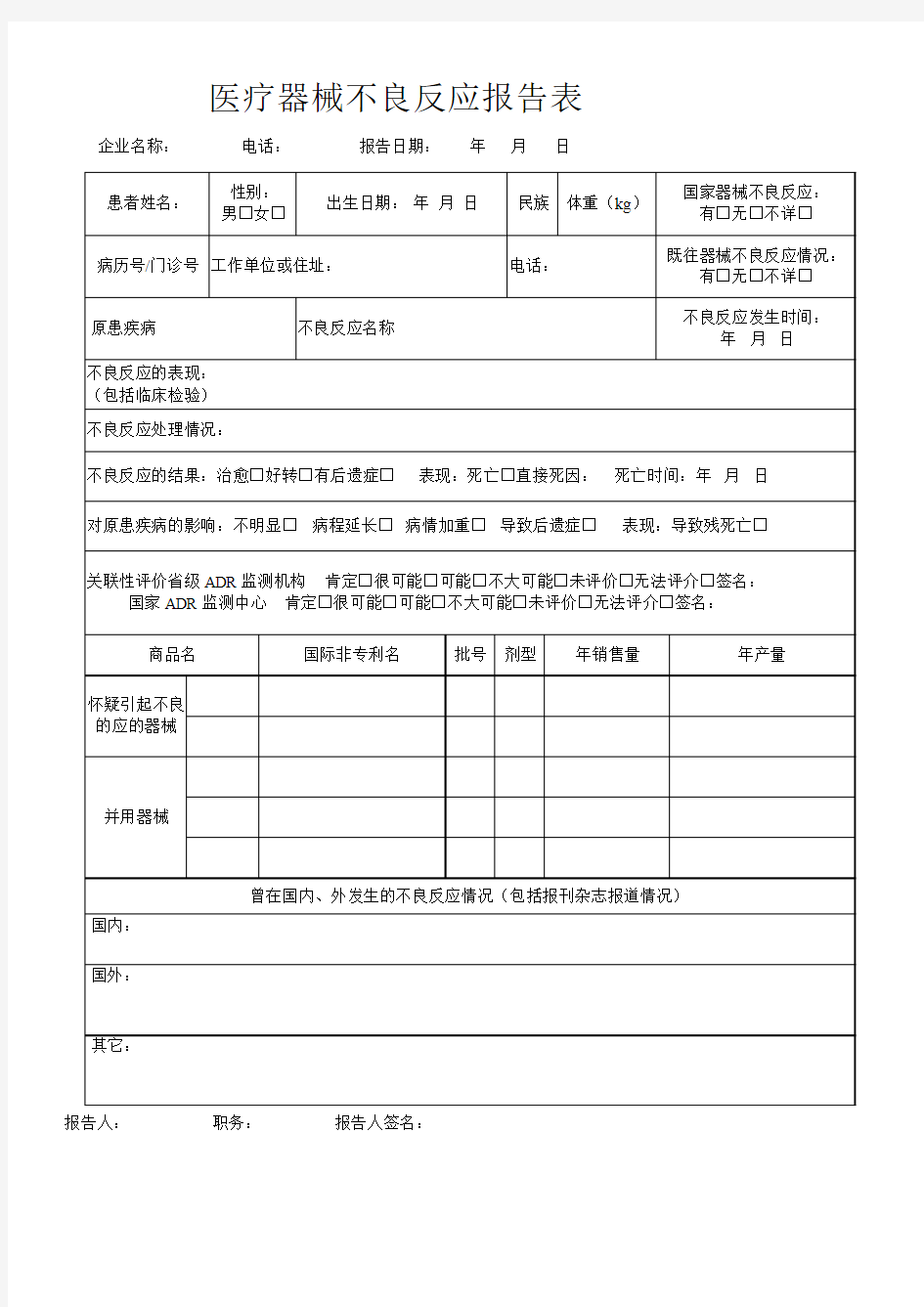
医疗器械不良反应报告表企业名称:电话:报告日期:年月日
报告人:职务:报告人签名:
药品不良反应/ 事件报告表 首次报告□跟踪报告□编码: 报告类型:新的□严重□一般□报告单位类别:医疗机构□经营企业□生产企业□个人□
药品不良反应/ 事件报告表示例 首次报告□跟踪报告□编码:
1.《药品不良反应/事件报告表》应填写真实事件,报表所列患者信息及怀疑药品信息项目必须真实、完整、准确。 2.《药品不良反应/事件报告表》填写字迹要清晰,其中选择项画“√”,叙述项应准确、简明,不得有缺漏项。 3. 新的□严重□一般□ (1)新的药品不良反应:是指药品说明书中未载明的不良反应。说明书中已有描述,但不良反应发生的性质、程度、后果或者频率与说明书描述不一致或者更严重的,按照新的药品不良反应处理。 根据不良反应/事件损害的严重程度,不良反应/事件可能是新的严重的,也可能是新的一般的。(2)严重药品不良反应,是指因使用药品引起以下损害情形之一的反应: 1) 导致死亡; 2)危及生命; 3)致癌、致畸、致出生缺陷; 4)导致显着的或者永久的人体伤残或者器官功能的损伤; 5)导致住院或者住院时间延长; 6)导致其他重要医学事件,如不进行治疗可能出现上述所列情况的。 (3)一般:指新的、严重的药品不良反应以外的所有不良反应。 4. 单位名称:必须填写单位的完整全称,如日照市人民医院。 5. 部门:应填写科室的标准全称,如:消化内科、普外三科等。 6. 电话:填写报告部门(即科室)的电话。 7. 报告日期:指上交不良反应/事件报告的时间。新的或严重的药品不良反应/事件应于发现之日起15日内报告,其中死亡病例须立即报告,其他药品不良反应应30日内报告。有随访信息的,应当及时报告。 8. 患者姓名:填写患者真实全名。 9. 体重:以千克为单位。如果不知道准确体重,请做一个最佳的估计。 10. 联系方式:最好填写患者的联系电话,也可填写患者的通信地址。 11. 家族药品不良反应/事件及既往药品不良反应/事件情况:请选择正确选项。如果选择“有”,则在报告的空白处清晰叙述。 12. 不良反应/事件名称:应填写不良反应中最主要、最明显的症状。 13. 不良反应/事件发生时间:应填写发生不良反应/事件的确切时间。 14. 病历(门诊)号:请如实填写,便于查找病例,具体分析不良反应/事件。 15. 不良反应/事件过程描述(包括症状、体征、临床检验等)及处理情况: 不良反应过程描述应具体、规范,须体现出“3个时间、3个项目和2个尽可能”。 *3个时间:①不良反应发生的时间;②采取措施干预不良反应的时间;③不良反应终结的时间; *3个项目:①第一次药品不良反应出现时的相关症状、体征和相关检查;②药品不良反应动态变化的相关症状、体征和相关检查;③发生药品不良反应后采取的干预措施结果;
医疗器械不良反应总结 医疗器械不良反应总结怎么写,以下是XX精心整理的相关内容,希望对大家有所帮助! 医疗器械不良反应总结20XX年上半年,全院共上报药品不良反应43例,医疗器械监测报表零报告。 一、医疗器械不良反应相关情况分析 根据《医疗器械不良反应监测和在评价管理办法(试行)》的工作要点,应坚持“可疑即报”原则,增加报告数量,以推广“医疗器械不良反应事件监测系统”为契机,大力提高报告质量,要求二级及二级以上医疗机构都要突破零报告,而我院医疗器械不良反应事件监测上报为零报告。结合我院实际情况分析,我院医疗器械监测报表为零报告的原因可能为以下几点: 1.医务人员对药品与医疗器械不良反应事件监测概念模糊,不能准确定义不良反应事件,导致漏报; 2. 科室隐瞒不报医疗器械不良反应事件,由于未造成不良后果,未引起科室重视并上报; 3. 没有医疗器械不良反应事件 针对这一情况,应进一步加强度医疗器械不良反应事件监测工作的组织领导,对医疗器械不良事件报告管理职责分别进行梳理和明确,要求各科室发现或知悉医疗器械不良事件后,在规定时限内及时上报,建立并保存医疗器械不良事
件监测记录;不断创新,进一步完善医疗器械不良反应事件监测体系;加强医务人员对药品与医疗器械不良反应事件监测概念的宣传,组织专家对医务人员进行相关医疗器械不良反应事件的培训,提高医务人员对医疗器械不良反应事件监测的上报意识。 二、药品不良反应报告情况分析 20XX年上半年药品不良反应监测报表共有43例,具体分布如下表: 根据医院开放床位数、年住院病人人次,医院每年应上报450例,而医院上半年仅上报43例,大部分科室只有1例,更有部分外科上半年为“零上报”,未能完成了药品不良反应的监测工作。 上报例数过少说明了我院药品不良反应监测工作中存在不少问题,主要是医务人员对“零上报”的理解不足,认为“零上报”就是不上报;然后是医务人员在发生药品不良反应时怕受惩罚,隐瞒不报;还有药品不良反应的上报机制不够完善,间接导致迟报或漏报。医院需进一步完善相关制度,狠抓落实相关措施,努力提高全院药械不良反应监测水平,即以下四个方面: 1、要加强宣传,开展多种形式的教育培训活动。对各科室进行宣传,不断提高他们对做好药品不良反应监测工作重要意义的认识。同时,要积极聘请专家就有关方面知识进
药品不良反应分析报告 (20 年月--20 年月) 生产企业:(盖章) 地址: 联系人: 电话: 报告日期: 一、企业监测体系建设概况
我司于2006年已建立药品不良反应报告和监测体系,并有效实施。2012年按《药品不良反应报告和监测管理办法》(卫生部令第81号)》和《浙江省药品不良反应报告和监测管理实施细则》完善我司药品不良反应监测体系、组织机构,明确其职责。 我司监管网络为一线销售人员从医院收集药品不良反应报告,汇总至市场部相关人员。市场部相关人员将数据汇总至相应QA人员,QA人员将数据上报至国家药品不良反应监测系统中。 组织机构: 我司处理药品不良反应/事件的专职部门为质量保证部,监测部门为市场部。 组成人员: 质量负责人、质保部负责人、一名市场部人员、一名QA人员。 职责: (1)市场部人员和QA人员及时收集与公司生产的药品有关的安 全性信息,发现与公司有关的药品不良反应,及时通过药品不良反应监测信息网络报告。每年向所在地药品不良反应监测机构提交药品不良反应监测工作报告; (2)对严重药品不良反应或者药品群体不良事件进行调查,必要时对药品采取紧急控制措施,如召回; (3)配合各级食品药品监督管理局、卫生行政部门和药品不良反应监测机构对药品不良反应或者群体不良事件的调查,并提供调查所需的资料,并填写《药品不良反应/事件报告表》,填报内容应真实、完整、准确。 (4)开展药品不良反应报告数据与药品质量的关联性研究,必要时进行重点监测或再评价;
(5)按要求撰写和提交定期安全性更新报告; (6)正确介绍药品的使用要求和注意事项等,将说明书修改等安全性信息及时告知相关药品经营企业和医疗机构。 (7)经常查阅国家食品药品监督管理局定期通报国家药品不良反应报告和监测情况。 二、公司品种概况 我司现有1个品种:****(批准文号:国药准字*****)。20**年1月1日至9月30日,我司对该品种进行了生产......... 无监测期品种和重点监测品种。 三、产品基本信息和不良反应收集情况 (一)不良反应反馈数据核实情况: 无。 (二)反馈数据涉及产品的基本信息: 产品的批准文号、通用名称、剂型、统计周期内的销量、不良反应报告例数、严重的不良反应例数及其构成比、新的一般的不良反应例数及其构成比、死亡病例数见附表1。 我司无文号转入或转出情况。 (三)其他不良反应信息收集情况: 在此次报告时间段内,我司未收集到到涉及本企业药品的不良反应或可疑不良反应信息情况。
《药品不良反应/事件报告表》填写要求 《药品不良反应报告和监测管理办法》第十三条明确规定:药品生产、经营企业和医疗卫生机构必须指定专(兼)职人员负责本单位生产、经营、使用药品的不良反应报告和监测工作,发现可能与用药有关的不良反应应详细记录、调查、分析、评价、处理,并填写《药品不良反应/事件报告表》,每季度集中向所在地的省、自治区、直辖市药品不良反应监测中心报告,其中新的或严重的药品不良反应应于发现之日起15日内报告,死亡病例须及时报告。 (一)填写注意事项: 1.《办法》第十四条规定:《药品不良反应/事件报告表》的填报内容应真实、完整、准确。 2.《药品不良反应/事件报告表》是药品安全性监测工作的重要档案资料,手工报表需要长期保存,因此需用钢笔、签字笔书写,填写内容、签署意见(包括有关人员的签字)字迹要清楚,不得用报告表中未规定的符号、代号、不通用的缩写形式和花体式签名。其中选择项画“√”,叙述项应准确、完整、简明,不得有缺漏项。 3.每一个病人填写一张报告表。 4.个人报告建议由专业人员填写,可以是诊治医务人员、药品生产、经营企业专(兼)职人员及专业监测机构人员。 5.尽可能详细地填写报告表中所要求的项目。有些内容无法获得时,填写“不详。 6.对于报告表中的描述性内容,如果报告表提供的空间不够,可另附A4纸说明。在纸的顶部注明“附件”,所有的附件应按顺序标明页码,附件中必须指出描述项目的名称。
7.补充报告:如需作补充报告时,请注意与原报表编号保持一致,并在报告左上方注明“补充报告”,与原报表重复的部分可不必再填写。补充报告也可不填写报告表,只需附纸说明补充内容即可,但须注明原报表编号、单位名称、补充报告时间、报告人。 (二)填写详细要求 1.新的、严重、一般: 新的ADR:是指药品说明书中未载明的ADR。 严重ADR:是指因服用药品引起以下损害情形之一的反应: ⑴引起死亡; ⑵致癌、致畸、致出生缺陷; ⑶对生命有危险并能够导致人体永久的或显著的伤残; ⑷对器官功能产生永久损伤; ⑸导致住院或住院时间延长。 一般的ADR:是指除新的、严重的ADR以外的所有ADR。 2.单位名称:填写医疗卫生机构、药品生产企业或经营企业的完整全称。如:“镇江市第一人民医院”,不可填“一院”。 3.部门:填写报告单位的具体报告部门, 应填写标准全称或简称,如:“普通外科二病房”或“普外二”,如连锁药店应填具体的门店,零售药店可填写药店名称。
药品不良反应/ 事件报告表 首次报告□ 跟踪报告□ 编码: 报告类型:新的□ 严重□ 一般□ 报告单位类别:医疗机构□ 经营企业□ 生产企业□ 个人□ 不良反应事件名称:不良反应事件发生时间:年月日不良反应/ 事件过程描述(包括症状、体征、临床检验等)及处理情况(可附页) 不良反应/ 事件的结果:痊愈□好转□ 未好转□ 不详□ 有后遗症□ 表现: 死亡□ 直接死因:死亡时间:年月日 停药或减量后,反应/ 事件是否消失或减轻?是□ 否□不明□ 未停药或未减量□再次使用可疑药品后是否再次出现同样反应/ 事件?是□ 否□ 不明□ 未再使用□
电子邮箱: 签名:××× 药 品 不 良 反 应 / 事 件 报 告 表示例 不良反应 / 事件过程描述(包括症状、体征、临床检验等)及处理情况(可附页) 一般格式为:患者因×××疾病于×××月×××日(必要时应详细到×××时分)以×××途径给予×××药品,×××剂量,用药×× ×时间出现×××反应(反应描述须明确、具体) ,×××时间后给予是否停药及×××处理(包括以×××途径给予×××药品及×××剂 和其他处理措施) ,处理后×××时间患者转归情况。 对原患疾病的影响: 不明显□ 病程延长□ 病情加重□ 导致后遗症□ 导致死亡□ 报告人评价: 肯定□ 很可能□ 可能□ 可能无关□ 待评价□ 无法评价□ 签名:××× 报告单位评价: 肯定□ 很可能□ 可能□ 可能无关□ 待评价□ 无法评价□ 签名:××× 报告人信息 患者姓名:××× 性别:男□女□ 出生日期: 年 月 日 或年龄: ×× 民族:×× 体重( kg ):×× 联系方式:×××××× 原患疾病:指患者此次入 诊的主要疾病(如果有多 疾病可以补充在相关重要 是备注里面),不能写字 院或就 种慢性 信息或 母缩写。 医院名称:三亚市中医院 病历号 / 门诊号:××××(务必 填写) 既往药品不良反应 / 事件:有□需提供药品通用名称及具体反应 无□ 不详□ 家族药品不良反应 / 事件:有□需提供药品通用名称及具体反应 无□ 不详□ 报告单位类别:医疗机构□ 其他□ 相关重要信息: 吸烟史□ 饮酒史□ 妊娠期□ 肝病史□ 肾病史□ 过敏史□此处是提供有否食物等过敏史 首次报告□ 跟踪报告□ 报告类型:新的□ 严重□ 一般□ 编码: 经营企业□ 生产企业□ 个人□ 其他□ 药 品 批准文号 商品 名称 通用名称 (含剂型) 生产厂 家 生产批号 用法用量 (次剂量、 途径、 日次数) 用药起止时 间 用药原因 怀 疑 药 品 国药准字 此处填写药品的 通用名 称。注射剂 包含注射液和粉 针剂,请认真选择 正确剂型 本次使用药 物的生产批 号 包括每次用药剂 量、 给药途径、 每日给药 次数, 例如,5mg , 口服, 每日 2 次。 指使用药品 的同 一剂量 的开始时 间 和停止时间 填写使用该药品的原因,应详 细填 写。例如:患者高血压病 史,此次因肺部感染而注射氨 苄青霉素引起不良反应,用药 原因栏应填写肺部 感染 并 用 药 品 同上 量, 不良反应 / 事件的结果:痊愈□ 死亡□ 好转□ 直接死因: 未好转□ 不详□ 有后遗症□ 表现: 死亡时间:× 年 × 月 × 日 停药或减量后,反应 / 事件是否消失或减轻? 再次使用可疑药品后是否再次出现同样反应 / 事件? 是□ 否□ 是□ 否□ 不明□ 不明□ 未停药或未减量□ 未再使用□ 关联性评价 联系电话:务必正确填写 职业:医生□ 药师□ 护士□ 其他□ 不良反应 / 事件名称:应填写不良反应中最主要、最明显的症状。 不良反应 / 事件发生时间:× 年 × 月× 日(应填写发生不良反应 / 事件 的确切时间)
产品质量跟踪和不良反应报告制度 1、目的:为了促进合理使用医疗器械,提高医疗器械的质量和医疗器械的效用水平,特制定本制度。 2、依据:《医疗器械监督管理条例》。 3、适用范围:适用于所有医疗器械的质量跟踪和不良反应报告的管理。 4、职责:全体员工对本制度的实施负责,质量管理员负责监督管理。 5、制度内容: 5.1、收集有关生产厂家的资料和医疗器械质量标准方面的资 料,经常与厂家保持联系,关注医疗器械生产质量的变化整理归档。 5.2、建立客户和医疗器械的质量档案,公司的质量管理人员要经常与客户保持联系,定期访问记录,并整理归档。 5.3、接到客户反映质量问题时,要高度重视,并立即派员到 该客户处了解情况,分析出现问题的原因,如果是由于使用不当造成的,要当场指出其错误之处,如果是商品本身的质量问题,则须按实际情况给予处理。 5.4、质量管理员负责本公司经营医疗器械不良反应的报告与组织管理工作。
5.5、各岗位人员应注意收集从本公司售出的医疗器械发生的不良反应的反馈情况,一旦发现,应及时向质量管理员报告。 5.6、质量管理员对收集反馈的医疗器械不良反应情况,要进行详细记录、调查,核实、汇总后,及时向当地食品药品监督管理局进行报告。 5.7、收集的医疗器械不良反应信息,应在当天反馈到质量管理员,以便核实上报。 5.8、对其中严重、罕见的新的医疗器械不良反应,须采用有效的方式快速报告,最迟不得超15 个工作日报告到省食品药品监督管理局。 不良事件报告记录 A.患者资料 1.患者姓名: 2 .年龄: 3.性别:□男□女 4.预期治疗疾病: 5.并发疾病: 6. 既往疾病: B .不良事件情况 7 .事件后果 □死亡(时间)□危胁生命 □残疾□出生缺陷□其它 8.事件发生日期:年月日
7.医疗器械不良事件分析总结 2017年医疗器械不良事件报告工作在全院临床及相关科室共同努力下,取得一定成果和进步。今年共计收到临床科室主动上报的医疗器械不良事件5例。2017年医疗器械不良事件的发生数量不多、严重程度不大,说明在不良事件的管理上,科室给予一定程度重视,加强了培训,以及我们医院所选用的医疗器械质量过硬,从而收到了一定效果。 一、不良事件统计: (一)护理类耗材:1、一次性使用无菌导尿包共0例;2、一次性使用无菌注射器(带针)共5例;3、一次性使用2腔无菌导尿管共0例;4、一次性使用3腔乳胶导尿管共0例;5、一次性使用无菌注射器共0例;6静脉留置针0例(二)透析及透析管路:共0例。 (三)其他医用耗材:1一次性使用心电电极共0例。2输液泵使用0例;2高频电刀使用0例;3麻醉机使用0例;4骨科钢板0例;5人工晶体0例;呼吸机使用0例 二、原因分析: 1、分析今年不良事件发生的原因,主要是由医疗器械的质量所造成。 2、临床科室使用时,不完全按照正确的操作流程操作也会造成不良事件的发生。 3、其他个别情况造成不良事件的发生,例如过敏体质的患者使用卫生耗材时发生过敏反应;医疗器械在搬运时发生碰撞,从而影响到使用等。 三、改进方法: 1、继续鼓励不良事件主动上报,发生不良事件后,及时积极与相关部门和人员合作,将患者的损害降到最低,最大限度的保障患者的生命安全。对于经常或者特别严重的医疗器械不良事件,要在医院会议上讨论,将个别经验教训作为教材,在全院引起重视。 2、加强重点人员的管理,加强年轻科室人员能力培训,规范各项操作规程,正确使用医疗器械。工作时做到井然有序,减少因操作不当引起的不良事件。 3、根据发生的问题从源头上、从流程制度上解决不良事件。同时,在选用医疗
医疗机构医疗器械不良事件报告举例 按照医疗器械分类目录对不同品种医疗器械的主要表 现进行了列举,供各医疗机构参考。 医疗器 分类名称产品名称不良事件表现械分类 6801基础外科手术器械 6804眼科手术器械 6805耳鼻喉科手术器械 6806口腔科手术器械医用缝合针 医用缝合针断裂 (不带线) 一次性使用 操作不利 基础外科用刀 备皮刀 手术刀片生锈、折断、缺损 基础外科用剪手术剪生锈、缺损 基础外科用钳 持针钳断裂、缺损 止血钳血管钳使用后有缺损 基础外科用镊 无损伤镊弹力不够 夹 一次性使用 盘底不平,凹槽太浅,导致消毒液外溢 基础外科其它换药盘 器械一次性使用 断裂划破医生手指、异物、边缘不整齐有倒刺压舌板 其他 一次性卫生 导致皮肤破损 垫 眼科手术用其常规眼科手 睫毛镊夹不住睫毛 他器械术器械包 耳鼻喉科用其一次性使用检查时弹簧弹出、闭合欠佳造成鼻腔窥见困他器械鼻镜难 口腔用镊、夹一次性镊子折断 口腔用其它器 械口腔用钩、根管锉针器械分离 针 一次性使用异物、器械盒封闭不严、断裂、生锈
口腔用其它器齿科器械盒 压舌板 械一次性使用毛刺、断裂 1/21
6808腹 部外科手其他 术器械 矫形(骨科) 外科用钳 矫形(骨科)6810矫外科用有源器形外科械 (骨科) 手术器械 其他 6812妇妇产科用剪产科用手妇产科用其他术器械器械 6815注 射穿刺器注射穿刺器械械一次性使用 不能击发、打开包装后,钉已经出仓、手柄开直线型吻合 裂 器 半月板篮钳篮钳头部断裂脱落于膝关节腔内 助力枪助力枪上的螺丝脱落 创伤手术工 钻孔错误 具 脊柱内固定 系统配套工工具断裂遗留在螺钉上 具 会阴剪螺丝脱落、刀刃有破损 阴道牵开器无螺丝 泵用注射器针头弯曲 一次性使用 无菌避光注注射乳头断裂 射器 一次性使用 动静脉瘘穿穿刺针有折痕 刺针 一次性使用 冠状动脉注漏液 射器 注射器乳头端断裂不能使用 包装盒内无针头 动脉血气针 包装封口密闭不严 动静脉穿刺 断裂、脱落、钢针偏斜 器 动脉采血器漏气、异物、漏血 一次性使用 针头外露,刺破针帽、无法推出余下液体、针无菌胰岛素 头脱落、断裂、针头堵塞 注射器 一次性使用 真空采血器包装破损、脱节、漏血配套 用针 一次性使用断裂、疼痛、脱落、裂口、外包装破损、穿刺静脉输液针针平头无尖 2/21
附表1 药品不良反应/ 事件报告表(书写模版)首次报告□跟踪报告□编码:
药品不良反应报告表 部分项目填报注意事项 一、药品不良反应事件名称及描述 1.如果患者出现皮疹伴瘙痒,不要把二者同时列为一个不良反应,应当分类描述为“皮疹;瘙痒”, 对于皮疹得发生部位、大约形态进行描述; 2.如果患者发生多种过敏反应,就不用分类描述,直接描述为“过敏反应”或就是“过敏样反应”;不属于过敏反应得其她症状,应当分类描述; 3.如果患者出现过敏性休克,就必须描述患者得临床表现(包含呼吸道阻塞、微循环障碍、中枢神经系统症状及皮肤过敏症状)及体征; 例如头晕、面色苍白、呼吸困难、胸闷、腹痛、出汗、脉搏增快及血压下降等;此时相应得体征进行描述,如体温、心率、血压、呼吸频率等;还包含不良反应发生前后得症状与体征得动态变化。 4.如果患者出现血象异常,要将不良反应发生前后相应指标、实验室检查进行描述; 例如患者白细胞降低,此时就需要提供患者入院时(或服药前)白细胞指数,服药后发生不良反应就是监测得白细胞指数以及采取措施停药后患者白细胞有所
恢复得指数。 5.如果患者出现消化道反应,例如腹泻、呕吐等,请具体描述一哈相关得症状; 例如腹泻,一日几次、什么性状;呕血,一日几次、颜色等性状;呕吐,一日几次、内容物就是什么。 二、药品不良反应发生后采取得措施及转归 1、药品不良反应发生以后,主要采取得治疗措施要进行描述。例如立即停药,给与抗过敏治疗(过敏反应)、给与升白细胞治疗(白细胞下降)、给与物理降温(高热)等对症治疗。具体得治疗措施,例如给与地塞米松10mg肌肉注射,要尽量详细描述对症治疗得药物及剂量。 2、药品不良反应得转归,要尽量描述采取对症治疗之后患者得转归。有得医疗机构在患者刚刚发生不良反应,采取措施尚未缓解得时候就立即上报,这种就是不规范得。 国家规定药品不良反应报告得上报时限。大家应当按照规定时限完整得对药品不良反应进行上报。
附件1: 《可疑医疗器械不良事件报告表》及填写要求 可疑医疗器械不良事件报告表 报告日期: 年 月 日 编 码: 报告来源: 生产企业 经营企业 使用单位 单位名称: 联系地址: 邮 编: 联系电话: 报告人签名: 国家食品药品监督管理局制 A .患者资料 1.姓名: 2.年龄: 3.性别 男 女 4.预期治疗疾病或作用: B .不良事件情况 5.事件主要表现: 6.事件发生日期: 年 月 日 7.发现或者知悉时间: 年 月 日 8. 医疗器械实际使用场所: 医疗机构 家庭 其他(请注明): 9.事件后果 死亡 (时间); 危及生命; 机体功能结构永久性损伤; 可能导致机体功能结构永久性损伤; 需要内、外科治疗避免上述永久损伤; 其他(在事件陈述中说明)。 10.事件陈述:(至少包括器械使用时间、使用目的、使用依据、 使用情况、出现的不良事件情况、对受害者影响、采取的治疗 措施、器械联合使用情况) 报告人: 医师 技师 护士 其他 C .医疗器械情况 11.产品名称: 12.商品名称: 13.注册证号: 14.生产企业名称: 生产企业地址: 企业联系电话: 15.型号规格: 产品编号: 产品批号: 16. 操作人: 专业人员 非专业人员 患者 其他(请注明): 17. 有效期至: 年 月 日 18.生产日期: 年 月 日 19. 停用日期: 年 月 日 20. 植入日期(若植入): 年 月 日 21. 事件发生初步原因分析: 22. 事件初步处理情况: 23.事件报告状态: 已通知使用单位 已通知生产企业 已通知经营企业 已通知药监部门 D.关联性评价 (1)使用医疗器械与已发生/可能发生的伤害事件之间是否具有合理的先后时间顺序? 是□ 否□ (2)已发生/可能发生的伤害事件是否属于所使用医疗器械可能导致的伤害类型?是□ 否□ 不清楚□ (3)已发生/可能发生的伤害事件是否可用合并用药和/或械的作用、患者病情或其他非医疗器械因素来解释? 是□ 否□ 不清楚□ 评价结论:很可能□可能有关□可能无关□无法确定□ E. 不良事件评价 24.省级监测技术机构评价意见(可另附附页): 25.国家监测技术机构评价意见(可另附附页):
管理制度编号:YTO-FS-PD991 医疗器械不良事件报告制度通用版 In Order T o Standardize The Management Of Daily Behavior, The Activities And T asks Are Controlled By The Determined Terms, So As T o Achieve The Effect Of Safe Production And Reduce Hidden Dangers. 标准/ 权威/ 规范/ 实用 Authoritative And Practical Standards
医疗器械不良事件报告制度通用版 使用提示:本管理制度文件可用于工作中为规范日常行为与作业运行过程的管理,通过对确定的条款对活动和任务实施控制,使活动和任务在受控状态,从而达到安全生产和减少隐患的效果。文件下载后可定制修改,请根据实际需要进行调整和使用。 一、医疗器械不良事件是指获准上市的、合格的医疗器械在正常使用情况下,发生的或可能发生的任何与医疗器械预期使用效果无关的有害事件。 二、医疗器械不良事件主要包括医疗器械已知和未知作用引起的副作用、不良反应及过敏反应等。副作用是指治疗使用的医疗器械所产生的与疾病防治目的无关的作用。 三、医务人员如发现可能与医疗器械有关的不良事件时,应及时报告不良事件专管员及科室负责人。 四、科室负责人发现或接到医疗器械不良事件报告后,应及时到现场察看,协助调查,并填写《医疗器械不良事件报告表》,核实后,提交医务部、护理部、院感科和设备科。特殊情况下,可先口头报告再补报书面材料。《医疗器械不良事件报告表》应包括患者情况、不良事件情况、医疗器械情况、初步处理情况等内容。 五、医务部、护理部、院感科和设备科接到医疗器械不良事件报告后,应联合组织调查,了解事件经过和相关
药品不良反应报告表 部分项目填报注意事项 一、药品不良反应事件名称及描述 1.如果患者出现皮疹伴瘙痒,不要把二者同时列为一个不良反应,应当分类描述为“皮疹;瘙痒”, 对于皮疹的发生部位、大约形态进行描述; 2.如果患者发生多种过敏反应,就不用分类描述,直接描述为“过敏反应”或是“过敏样反应”;不属于过敏反应的其他症状,应当分类描述; 3.如果患者出现过敏性休克,就必须描述患者的临床表现(包含呼吸道阻塞、微循环障碍、中枢神经系统症状及皮肤过敏症状)及体征; 例如头晕、面色苍白、呼吸困难、胸闷、腹痛、出汗、脉搏增快及血压下降等;此时相应的体征进行描述,如体温、心率、血压、呼吸频率等;还包含不良反应发生前后的症状和体征的动态变化。 4.如果患者出现血象异常,要将不良反应发生前后相应指标、实验室检查进行描述; 例如患者白细胞降低,此时就需要提供患者入院时(或服药前)白细胞指数,服药后发生不良反应是监测的白细胞指数以及采取措施停药后患者白细胞有所恢复的指数。
5.如果患者出现消化道反应,例如腹泻、呕吐等,请具体描述一哈相关的症状; 例如腹泻,一日几次、什么性状;呕血,一日几次、颜色等性状;呕吐,一日几次、内容物是什么。 二、药品不良反应发生后采取的措施及转归 1、药品不良反应发生以后,主要采取的治疗措施要进行描述。例如立即停药,给与抗过敏治疗(过敏反应)、给与升白细胞治疗(白细胞下降)、给与物理降温(高热)等对症治疗。具体的治疗措施,例如给与地塞米松10mg肌肉注射,要尽量详细描述对症治疗的药物及剂量。 2、药品不良反应的转归,要尽量描述采取对症治疗之后患者的转归。有的医疗机构在患者刚刚发生不良反应,采取措施尚未缓解的时候就立即上报,这种是不规范的。 国家规定药品不良反应报告的上报时限。大家应当按照规定时限完整的对药品不良反应进行上报。 备注:提供一个药品不良反应/事件报告表填写模板(见附表),供大家参考。
医疗器械不良事件分析总结-标准化文件发布号:(9556-EUATWK-MWUB-WUNN-INNUL-DDQTY-KII
医疗器械不良事件分析总结2013年医疗器械不良事件报告工作在全院临床及相关科室共同努力下,取得一定成果和进步。今年共计收到临床科室主动上报的医疗器械不良事件12例。2013年医疗器械不良事件的发生数量不多、严重程度不大,说明在不良事件的管理上,科室给予一定程度重视,加强了培训,以及我们医院所选用的医疗器械质量过硬,从而收到了一定效果。 一、不良事件统计:(一)护理类耗材:1、一次性使用无菌导尿包共4例; 2、一次性使用无菌注射器(带针)共2例; 3、一次性使用无菌导尿管共2例; 4、一次性使用乳胶导尿管共1例; 5、一次性使用无菌注射器共1例。(二)透析及透析管路:1、穿刺针(德朗)共1例。(三)其他周边医用耗材:1一次性使用心电电极共1例。 二、原因分析:1、分析今年不良事件发生的原因,主要是由医疗器械的质量所造成。2、临床科室使用时,不完全按照正确的操作流程操作也会造成不良事件的发生。3、其他个别情况造成不良事件的发生,例如过敏体质的患者使用卫生耗材时发生过敏反应;医疗器械在搬运时发生碰撞,从而影响到使用等。 三、改进方法:1、继续鼓励不良事件主动上报,发生不良事件后,及时积极与相关部门和人员合作,将患者的损害降到最低,最大限度的保障患者的生命安全。对于经常或者特别严重的医疗器械不良事件,要在医院会议上讨论,将个别经验教训作为教材,在全院引起重视。2、加强重点人员的管理,加强年轻科室人员能力培训,规范各项操作规程,正确使用医疗器械。工作时做到井然有序,减少因操作不当引起的不良事件。3、根据发生的问题从源头上、从流程制度上解决不良事件。同时,在选用医疗器械时,要提供符合安全标准的器具、材料或耗材。减少或者避免同类的医疗器械不良事件的发生。 2
药品不良反应/ 事件报告表 ※新的□严重□一般□医疗卫生机构□生产企业经营企业□个人□编码□□□□□□□□□□□□□□□□□□□单位名称:部门:※电话:※报告日期:年月日 ※电话: ◇不良反应/事件分析
※停药或减量后,反应/事件是否消失或减轻?是□否□不明□未停药或未减量□ ※再次使用可疑药品后是否再次出现同样反应/事件?是□否□不明□未再使用□ PS: 用法用量: 用量:___(毫升、微升、毫克、微克、粒、片、等……) 给药途径:____(静滴、肌注、皮下注射、口服、含服、外用等……) 不良反应/事件需填写具体症状,如: 皮肤反应:皮疹、斑丘疹、皮炎、荨麻疹、风团、水疱、瘙痒、面部水肿、眼睑水肿、皮肤红肿、硬结、局部发热、皮肤发冷、出汗、出血点、皮肤潮红等 胃肠道反应:恶心、反胃、反酸、呕吐、腹痛、腹胀、腹泻、大便次数增加、胃不适、便秘、便血、口干、口(舌)麻木、口腔金属味、消化道出血、胃溃疡、大小便失禁等 全身系统:胸闷、发热、寒战、颤抖、抖动、乏力、食欲不振等 神经系统:头晕、头痛、麻木、抽搞、癒痛、感觉异常、意识模糊、意识丧失、晕厥、牙关紧闭、反应迟钝、嗜睡、兴奋、坐立不安、烦躁、幻觉等 心血管系统:心慌、心悸、心律失常、心电图异常、心区不适、心动过缓、心脏停搏、紫绀、血压降低、血压升高、休克等 呼吸系统:呼吸困难、呼吸急促、咽喉不适、喉头水肿、咽喉发紧、喉部窒息感、舌水肿、哮喘等 肾功能:肾功能异常、无尿、少尿、排尿困难、肾衰竭等 肝功能:肝功能异常、转氨酶升高、黄疸、肝功能衰竭等。 其他:静脉炎、注射部位疼痛、红肿、硬结;牙龈出血、鼻出血(主要针对抗凝血药和容易导致出血的药面色苍白、月经紊乱、耳鸣、味觉异常、肌肉疼痛、骨髓抑制(血小板、白细胞、粒细胞等减少)、 肌肉痛、胸痛、腰痛等。 皮肤及其附件损害:渗出性红斑、多形性红斑、剥脱性皮炎 ?胃肠系统损害:消化性溃疡、胃肠道出血、呕血、肠梗阻 ?呼吸系统损害:支气管痉挛、哮喘、呼吸抑制、呼吸困难、喉水肿、肺水肿 ?中枢及外周神经系统损害:锥体外系病、张力亢进、语言障碍、舌麻痹、脑水肿、惊厥、昏迷 ?红细胞异常:溶血性贫血、全血细胞减少、骨髓抑制 ?心率及心律紊乱:心脏停搏、房颤 ?心血管系统一般损害:紫绀、体位性低血压、高血压、低血压 ?白细胞和网状内皮系统异常:粒细胞减少、白细胞减少 ?心外血管损害:脑出血、过敏性紫癜 ?肝胆系统损害:肝炎、肝细胞损害、肝功能异常 ?神经紊乱:谵妄、意识模糊 ?血小板和出血,凝血障碍:紫癜、血小板减少性紫癜、血小板减少、凝血障碍、非特异性出血 ?泌尿系统损害:血尿、肾功能异常、排尿困难、尿潴留、急性肾衰竭 ?代谢和营养障碍:痛风、高血糖、高尿酸血症、低血糖昏迷、低血糖、低血钾、低钠血症 ?肌肉骨骼系统损害:肌痛、肌病、横纹肌溶解
v1.0 可编辑可修改 附件1: 《可疑医疗器械不良事件报告表》及填写要求 可疑医疗器械不良事件报告表 报告日期:年月日编码: 报告来源:生产企业经营企业使用单位单位名称: 联系地址: 邮编:联系电话:A.患者资料 1.姓名:2.年龄: 3.性别男女 4.预期治疗疾病或作用: B.不良事件情况 5.事件主要表现: 6.事件发生日期:年月日 7.发现或者知悉时间:年月日 8. 医疗器械实际使用场所: 医疗机构家庭其他(请注明): 9.事件后果 死亡(时间); 危及生命; 机体功能结构永久性损伤; 可能导致机体功能结构永久性损伤; 需要内、外科治疗避免上述永久损伤; 其他(在事件陈述中说明)。
v1.0 可编辑可修改 报告人:医师技师护士其他16. 操作人:专业人员非专业人员患者其他(请注明): 23.事件报告状态: 已通知使用单位已通知生产企业 已通知经营企业已通知药监部门
报告人签名: 国家食品药品监督管理局制 填写要求 《可疑医疗器械不良事件报告表》由题眉、患者资料、不良事件情况、医疗器械情况、关联性评价、不良事件评价及题末7部分组成。 1.题眉 A.报告日期:是指填报人填报该次不良事件时的确切时间。 B.编码:由省(区、市)医疗器械不良事件监测技术机构填写,按以下排列方式: 省(区、市)年份流水号 □□□□□□□□□□□ 注:省(区、市)编码按中华人民共和国行政区划代码填写。在医疗器械不良事件监测系统中,编码由系统自动生成。 C.报告来源:是指填报可疑医疗器械不良事件单位的类别,填写时请选择相应的选项,并在“□”中划“√”。 D.单位名称:是指填报可疑医疗器械不良事件单位的全称,不可用简称。 E.联系地址、电话及邮编:是指填报可疑医疗器械不良事件单位的联系地址、电话及邮编。 2.患者资料 A.患者姓名:是指患者真实全名。若患者姓名无法获知,应填写未知;新生儿无姓名,应填写××子或××女。 B.年龄:是指患者发生可疑医疗器械不良事件时的实际年龄,字体为阿拉伯数字。若患者年龄小于1岁,应填写具体的月份或天数,如6个月。 C.性别:是指患者的性别,填写时请选择相应的选项,并在“□”中划“√”。 D.预期治疗疾病或作用:是指涉及不良事件的医疗器械用于治疗的疾病或者预计使用该医疗器械所发挥的作用,例如血管内支架用于治疗急性心肌梗死。 3.不良事件情况 A.事件主要表现:是指使用医疗器械后引发的、可能与该医疗器械使用有关的有害事件(且与质量、医疗事故无关)。填写不良事
制表单位: 紧急□ 一般√ 编号:□□□□□□□□□□□□□□□ 药品不良反应报告表(医疗单位使用) 医院名称:解放军第251医院科别:呼吸科电话:8785120 报告日期:2004年1月18日 患者姓名:高凤荣性别:男□女√ 出生日期:1967年4月2日民族:汉体重:56 (kg)家族药品不良反应:有□无□不详√ 病历号:235674 (门诊号)工作单位或地址:张家口市二毛纺厂电话:8161684 既往药品不良 反应情况:有√ 无□ 不详□ 原患疾病:慢性气管炎急性发作不良反应 名称: 速发性哮喘反应不良反应发生时间:2004年1月19日 不良反应的表现: (包括临床检验) 因慢性气管炎急性发作,抗感染治疗及雾化吸入后,症状消失,用5%葡萄糖注射液 500ml加黄芪注射液30ml静滴,用药2min,出现胸闷、呼吸困难、气喘、张口呼吸,查双肺 满布哮鸣音,考虑为黄芪注射液致速发性哮喘反应。 不良反应处理情况: 立即停止静滴黄芪注射液,吸氧,给予地塞米松注射液10mg,3min后,呼吸困难、气喘改善,2h后症状消失,双肺呼吸音清,无哮鸣音。
药品名称生产厂家批号剂型用药途径日剂量用药起止时间用药原因 怀疑引起不良反应的药品 黄芪注射液石家庄市神威药业有限公司0310621 注射液静滴 30ml 2004年1月17日 10:00-10:02 慢性气管炎急性发作 并用药品 5%葡萄糖注射液251医院制剂中心031107 灭菌溶液静滴500ml 2004年1月17日 10:00-10:02 溶媒 不良反应的结果:治愈√好转□有后遗症□ 表现:死亡□ 直接原因:死亡时间:年月日 对原患疾病的影响:不明显√病程延长□ 病情加重□导致后遗症□ 表现: 导致死亡□ 不良反应分析 1.用药与不良反应的出现有无合理的时间关系?有√ 无□ 2.反应是否符合该药已知的不良反应类型?是√否□不明□ 3.停药或减量后,反应是否消失或减轻?是√ 否□ 不明□ 未停药或未减量□ 4.再次使用可疑药品后是否再次出现同样反应?是□ 否□ 不明□ 未再使用√
附表1 药品不良反应/事件报告表 首次报告□跟踪报告□编码: 报告类型:新的□严重□一般□报告单位类别:医疗机构□经营企业□生产企业□个人□其他□ 患者姓名:性别:男□女□出生日期:年月日 或年龄: 民族:体重(kg):联系方式: 原患疾病:医院名称: 既往药品不良反应/事件:有□无□不详□ 病历号/门诊号:家族药品不良反应/事件:有□无□不详□ 相关重要信息:吸烟史□饮酒史□妊娠期□肝病史□肾病史□过敏史□其他□ 药品批准文号商品名称 通用名称 (含剂型) 生产厂家生产批号用法用量 (次剂量、途径、日次 数) 用药起止时间用药原因 怀 疑 药 品 并 用 药 品 不良反应/事件名称:不良反应/事件发生时间:年月日 不良反应/事件过程描述(包括症状、体征、临床检验等)及处理情况(可附页): 不良反应/事件的结果:痊愈□好转□未好转□不详□有后遗症□表现: 死亡□直接死因:死亡时间:年月日 停药或减量后,反应/事件是否消失或减轻?是□否□不明□未停药或未减量□ 再次使用可疑药品后是否再次出现同样反应/事件?是□否□不明□未再使用□ 对原患疾病的影响:不明显□病程延长□病情加重□导致后遗症□导致死亡□ 关联性评价报告人评价:肯定□很可能□可能□可能无关□待评价□无法评价□签名:报告单位评价:肯定□很可能□可能□可能无关□待评价□无法评价□签名: 联系电话:职业:医生□药师□护士□其他□ 报告人信息 电子邮箱:签名: 报告单位信息单位名称:联系人:电话:报告日期:年月日 生产企业请 填写信息来源 医疗机构□经营企业□个人□文献报道□上市后研究□其他□备注
严重药品不良反应,是指因使用药品引起以下损害情形之一的反应: 1)导致死亡; 2)危及生命; 3)致癌、致畸、致出生缺陷; 4)导致显著的或者永久的人体伤残或者器官功能的损伤; 5)导致住院或者住院时间延长; 6)导致其他重要医学事件,如不进行治疗可能出现上述所列情况的。 新的药品不良反应:是指药品说明书中未载明的不良反应。说明书中已有描述,但不良反应发生的性质、程度、 后果或者频率与说明书描述不一致或者更严重的,按照新的药品不良反应处理。 报告时限 新的、严重的药品不良反应应于发现或者获知之日起15日内报告,其中死亡病例须立即报告,其他药品不良反应30日内报告。有随访信息的,应当及时报告。 其他说明 怀疑药品:是指患者使用的怀疑与不良反应发生有关的药品。 并用药品:指发生此药品不良反应时患者除怀疑药品外的其他用药情况,包括患者自行购买的药品或中草药等。 用法用量:包括每次用药剂量、给药途径、每日给药次数,例如,5mg,口服,每日2次。 报告的处理 所有的报告将会录入数据库,专业人员会分析药品和不良反应/事件之间的关系。根据药品风险的普遍性或者严重程度,决定是否需要采取相关措施,如在药品说明书中加入警示信息,更新药品如何安全使用的信息等。在极少数情况下,当认为药品的风险大于效益时,药品也会撤市。
医疗器械不良事件监测及报告制度 一、医疗器械不良事件是指:获准上市的、合格的医疗器械在正常使用情况下,发生任何与医疗器械预期使用效果无关的有害事件。 二、医疗器械不良事件的监测是指对可疑医疗器械不良事件的发现、报告、评价和控制的过程。医疗器械不良事件监测工作,是预防医疗器械不良事件重复发生和蔓延,保证人民生命安全的重要工作。 三、为加强医疗器械的安全监管,规范医疗器械不良事件报告管理,发现可疑不良事件及时处置,并按规定上报,保障医疗器械临床使用安全,根据《医疗器械监督管理条例》及《昆明市医疗机构医疗器械质量管理规范》的相关规定,制定本制度。 四、医院成立医疗器械不良事件监测领导小组,完善管理制度,组织落实上级有关法律法规的培训工作,指导医院医疗器械不良事件监测工作的开展,监督、检查,确保医疗器械使用安全有效。 五、临床科室健全完善监测体系。科主任、护士长为科室医疗器械不良事件监测的负责人,指定专人做好医疗器械使用的详细登记,并对不良事件的信息进行收集,整理、上报。 六、报告医疗器械不良事件应当遵循可疑即报的原则。 七、医院各临床科室、门诊在诊疗过程活动中如发现医疗器械不良事件时,应立即停止使用,封存,向医疗器械管理科室报告。
八、医疗器械管理科室接到报告后应及时安排相关人员开展工作,对不良事件进行调查、分析、评价,在事件发生24小时内向省市药监局进行网络直报,填写《医疗器械不良事件报告表》交医院及卫生局主管部门,不得擅自处理。 九、根据不良事件的调查情况,医疗器械管理科室应及时向院内各相关科室通报,以引起警惕,避免造成新的伤害。 十、临床科室如对医疗器械不良事件隐瞒不报,经查实后根据情节轻重进行处罚。
附表1 药品不良反应 / 事件报告表(书写模版)首次报告□跟踪报告□编码:
药品不良反应报告表 部分项目填报注意事项 一、药品不良反应事件名称及描述 1.如果患者出现皮疹伴瘙痒,不要把二者同时列为一个不良反应,应当分类描述为“皮疹;瘙痒”, 对于皮疹的发生部位、大约形态进行描述; 2.如果患者发生多种过敏反应,就不用分类描述,直接描述为“过敏反应”或是“过敏样反应”;不属于过敏反应的其他症状,应当分类描述; 3.如果患者出现过敏性休克,就必须描述患者的临床表现(包含呼吸道阻塞、微循环障碍、中枢神经系统症状及皮肤过敏症状)及体征; 例如头晕、面色苍白、呼吸困难、胸闷、腹痛、出汗、脉搏增快及血压下降等;此时相应的体征进行描述,如体温、心率、血压、呼吸频率等;还包含不良反应发生前后的症状和体征的动态变化。 4.如果患者出现血象异常,要将不良反应发生前后相应指标、实验室检查进行描述; 例如患者白细胞降低,此时就需要提供患者入院时(或服药前)白细胞指数,
服药后发生不良反应是监测的白细胞指数以及采取措施停药后患者白细胞有所恢复的指数。 5.如果患者出现消化道反应,例如腹泻、呕吐等,请具体描述一哈相关的症状; 例如腹泻,一日几次、什么性状;呕血,一日几次、颜色等性状;呕吐,一日几次、内容物是什么。 二、药品不良反应发生后采取的措施及转归 1、药品不良反应发生以后,主要采取的治疗措施要进行描述。例如立即停药,给与抗过敏治疗(过敏反应)、给与升白细胞治疗(白细胞下降)、给与物理降温(高热)等对症治疗。具体的治疗措施,例如给与地塞米松10mg肌肉注射,要尽量详细描述对症治疗的药物及剂量。 2、药品不良反应的转归,要尽量描述采取对症治疗之后患者的转归。有的医疗机构在患者刚刚发生不良反应,采取措施尚未缓解的时候就立即上报,这种是不规范的。 国家规定药品不良反应报告的上报时限。大家应当按照规定时限完整的对药品不良反应进行上报。