
轻松实现域名解析本地化
每一个域名都对应一个IP地址,当你在浏览器中输入域名,实际上系统先要将此域名转换成IP地址,然后进行访问。而进行域名转换就需要用到DNS(Domain Name Server)域名系统,其主要工作就是寻找Internet域名并将它转换为IP地址。
有时候我们会遇到这样的问题,使用域名不能访问,而输入IP地址可以访问,这就是DNS 服务器出了问题。实际上我们可以建立一个本地域名解析系统,这样可以免去域名解析的时间,从而加快浏览的速度。
手工法
本地域名解析是通过“Hosts”文件来实现的。在Windows 98下,该文件在系统安装的目录下(一般为C:Windows);而在Windows 2000下,该文件在C:WINNTsystem32driversetc下。你可以利用记事本或其他编辑软件将其打开进行编辑。要注意的是,该文件是没有扩展名的。
编辑该文件要按照一定的格式,其格式为:
IP地址域名别名#注释。
你可以按照此格式添加到“Hosts”文件中,例如:211.99.206.167 https://www.doczj.com/doc/5f1809609.html, ccid #赛迪网。IP地址的获得可以利用ping命令来实现。使用该命令最好切换到MS-DOS窗口,命令的格式为:ping域名。例如ping https://www.doczj.com/doc/5f1809609.html,,回车后会得到这样的信息Pinging https://www.doczj.com/doc/5f1809609.html, [211.99.206.167] with 32 bytes of data,其中211.99.206.167就是其IP地址。别名的作用可以简化输入过程,你不需要输入整个域名,而只需要简单地输入别名,就可以访问该别名对应的站点,当然你也可以不定义别名。
在“Hosts”文件中,每一个域名占用一行,你只需要逐个添加就可以了。需要注意的是,如果你是用记事本建立这个文件,其保存的文件默认为文本文件,你需要改为无扩展名的文件。而在Windows系统下,默认是不显示常用文件的扩展名,你需要到文件管理窗口中选定“查看/文件”,在接下来的窗口中选择“查看”栏,将其中“隐藏已知文件类型的扩展名”前面的小勾去掉,这样你就可以看到文件的扩展名了。或者到MS-DOS窗口中,用REN命令来修改,所以说有时候DOS命令还是很有用的。
软件法
手工法虽然简单,但是太麻烦,如果域名太多,一个个地ping,还要编辑,真是累。所以对于我这种懒人,总会找些简单的办法。能够解决这个问题的软件很多,下面就给大家介绍两个。
一、FastNet99
FastNet99能够让使用者将IP地址、网址信息直接输进Windows目录底下的Hosts文件,当然也可以让使用者随时自行加入常用到的网址到原有的清单。另外,它还可以扫描IE收藏夹、Netscpae的书签并自动加入清单。
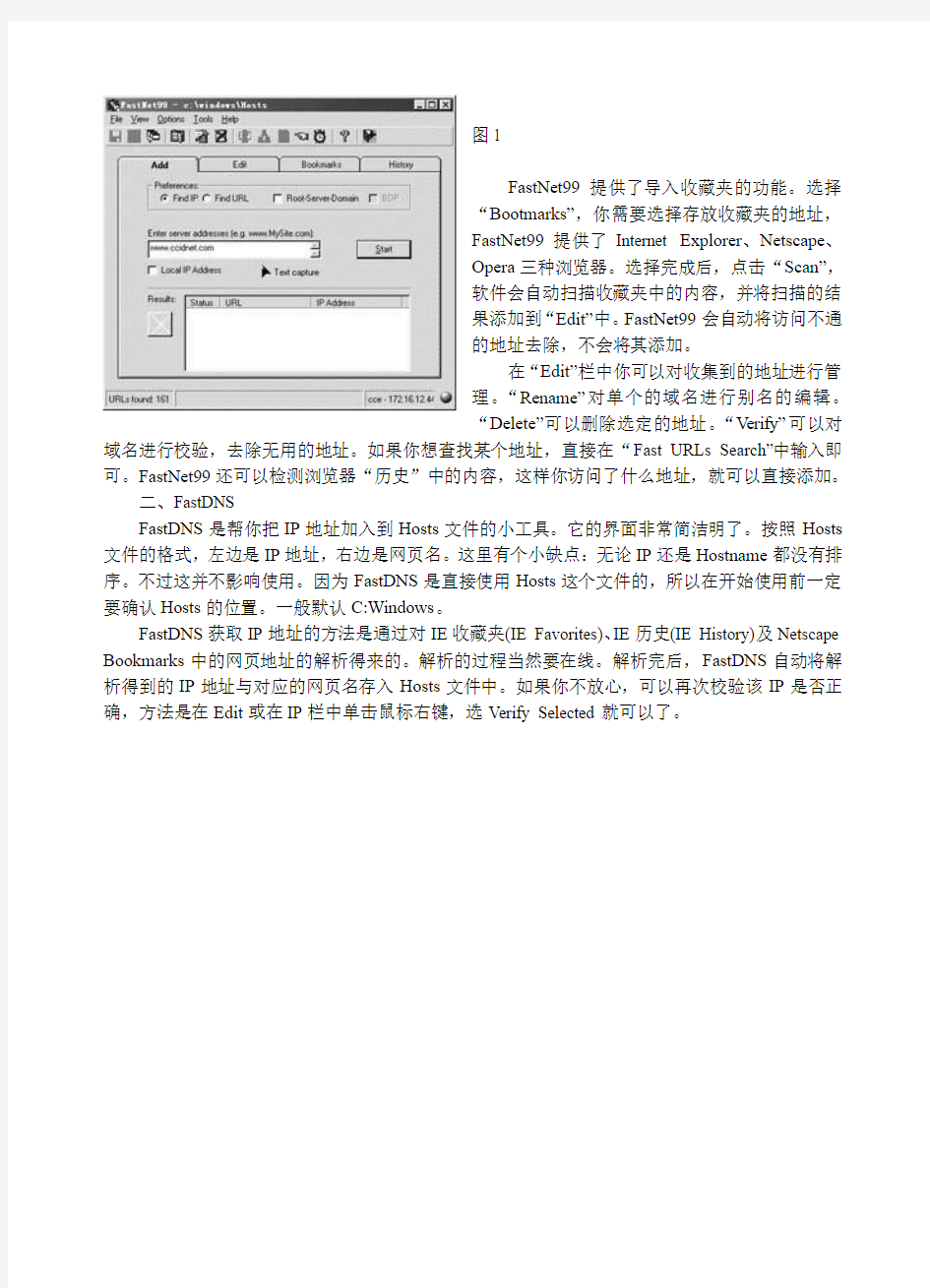
软件运行后,界面如图1。在“Add”栏中你可以输入需要解析的域名。首先在“Preferences”中选择“Find IP”,然后在“Enter Server addresse s”中输入域名,点击“Start”即可。这时你切换到“Edit”栏目,你会发现刚才输入的域名已经保存到该栏目中了。如果你觉得这样逐个输入太麻烦,在“Enter Server addresses”可以输入多个域名,需要通过“Ctrl+Enter”来换行。还有更简单的办法,在“Preferences”中选择“3 fields entry mode”,然后在“Root”和“Domain”中输入域名前缀和后缀,这样你只需要在“Server name”域名的中间部分就好了。完成后要记得保存哦!
图1
FastNet99提供了导入收藏夹的功能。选择
“Bootmarks”,你需要选择存放收藏夹的地址,
FastNet99提供了Internet Explorer、Netscape、
Opera三种浏览器。选择完成后,点击“Scan”,
软件会自动扫描收藏夹中的内容,并将扫描的结
果添加到“Edit”中。FastNet99会自动将访问不通
的地址去除,不会将其添加。
在“Edit”栏中你可以对收集到的地址进行管
理。“Rename”对单个的域名进行别名的编辑。
“Delete”可以删除选定的地址。“Verify”可以对域名进行校验,去除无用的地址。如果你想查找某个地址,直接在“Fast URLs Search”中输入即可。FastNet99还可以检测浏览器“历史”中的内容,这样你访问了什么地址,就可以直接添加。
二、FastDNS
FastDNS是帮你把IP地址加入到Hosts文件的小工具。它的界面非常简洁明了。按照Hosts 文件的格式,左边是IP地址,右边是网页名。这里有个小缺点:无论IP还是Hostname都没有排序。不过这并不影响使用。因为FastDNS是直接使用Hosts这个文件的,所以在开始使用前一定要确认Hosts的位置。一般默认C:Windows。
FastDNS获取IP地址的方法是通过对IE收藏夹(IE Favorites)、IE历史(IE History)及Netscape Bookmarks中的网页地址的解析得来的。解析的过程当然要在线。解析完后,FastDNS自动将解析得到的IP地址与对应的网页名存入Hosts文件中。如果你不放心,可以再次校验该IP是否正确,方法是在Edit或在IP栏中单击鼠标右键,选Verify Selected就可以了。
Blast 2.4.0+本地化详细流程(基于Windows系统) 1.程序获得。从NCBI上下载Blast本地化程序,下载地址: ftp://https://www.doczj.com/doc/5f1809609.html,/blast/executables/blast+/LATEST/ 64×安装版▲ 64×解压(绿色)版▲ 最好安装或解压到X盘根目录:如X:\blast,尽量简短,方便后边命令输入。 2.原始序列获得。方法1:找到转录组测序数据unigene数据库文件:unigene.fasta 或unigene.fa,若为unigene.fa则直接改后缀为.fasta即可。找到或修改后将数据库文件移动至Blast本地化程序目录“X:\blast\bin”。方法2:从NCBI中的ftp 库下载所需要库,链ftp://https://www.doczj.com/doc/5f1809609.html,/blast/db/FASTA/,其中nr.gz为非冗余的数据库,nt.gz为核酸数据库,month.nt.gz为最近一个月的核酸序列数据。下载的month.nt.gz先用WINRAR解压缩,然后用makeblastdb.exe格式化。方法3:利用新版blast自带的update_blastdb.pl进行下载,这需要安装perl程序。 注释:上述三种方法各有优缺点,前两种下载速度较快,但是每次进行检索都需要对数据库进行格式化(转化成二进制数据),第三种方法下载速度较慢,但是NCBI 中已经格式化好的,在进行本地检索时不需再进行格式化,直接用即可。 3.用文本编辑器(txt文件改名字及后缀)创建一个ncbi.ini文件,文件包含下 面内容:[NCBI]Data="C:\blast\data\" 先新建TXT文件,然后改属性,将ncbi.ini文件存放到C:\Windows 4.将Blast本地化程序目录添加路径中(该步骤非必须,但会给以后的操作带来 方便),方法: a)右击我的电脑选择属性,选择高级,点击环境变量,设置环境变量 b)系统变量中,选择Path,点击“编辑”,在变量值的后面添加Blast本地化 程序所在路径,E:\blast 点击确定,将安装路径添加到path。 5.运行MS-DOC。打开DOC窗口(点击开始,选择运行,打开的输入框中输 入“CMD”,确定),访问Blast本地化程序所在文件夹,依次输入:(1)X: 回车;(2)cd blast\bin,回车。
N C B I在线B L A S T使用方法与结果详解 IMB standardization office【IMB 5AB- IMBK 08- IMB 2C】
N C B I在线B L A S T使用方法与结果详解 BLAST(BasicLocalAlignmentSearchTool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。BLAST程序能迅速与公开数据库进行相似性序列比较。BLAST结果中的得分是对一种对相似性的统计说明。 BLAST采用一种局部的算法获得两个序列中具有相似性的序列。 Blast中常用的程序介绍: 1、BLASTP是蛋白序列到蛋白库中的一种查询。库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。 2、BLASTX是核酸序列到蛋白库中的一种查询。先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。 3、BLASTN是核酸序列到核酸库中的一种查询。库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。 4、TBLASTN是蛋白序列到核酸库中的一种查询。与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。 5、TBLASTX是核酸序列到核酸库中的一种查询。此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。 NCBI的在线BLAST: 下面是具体操作方法 1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。不同的blast程序上面已经有了介绍。这里以常用的核酸库作为例子。 2,粘贴fasta格式的序列。选择一个要比对的数据库。关于数据库的说明请看NCBI在线blast数据库的简要说明。一般的话参数默认。 3,blast参数的设置。注意显示的最大的结果数跟E值,E值是比较重要的。筛选的标准。最后会说明一下。 4,注意一下你输入的序列长度。注意一下比对的数据库的说明。 5,blast结果的图形显示。没啥好说的。 6,blast结果的描述区域。注意分值与E值。分值越大越靠前了,E值越小也是这样。7,blast结果的详细比对结果。注意比对到的序列长度。评价一个blast结果的标准主要有三项,E值(Expect),一致性(Identities),缺失或插入(Gaps)。加上长度的话,就有四个标准了。如图中显示,比对到的序列长度为1405,看Identities这一值,才匹配到1344bp,而输入的序列长度也是为1344bp(看上面的图),就说明比对到的序列要长一
本地blast的详细用法 Posted on 03 四月 2009 by 柳城,阅读 9,626 本地blast的详细使用方法 blast all -p blastn -i myRNA.fasta -d humanRNA.fasta -o myresult.blastout -a 2 -F F -T T -e 1e-10 解释如下: blastall: 这是本地化/命令行执行blast时的程序名字!(Tips:blastall直接回车就会给出你所有的参数帮助,但是英文的) -p: p 是program的简写,program在计算机领域中是程序的意思。此参数是指定要使用何种子程序,所谓子程序,就是针对不同的需要,如核酸序列和核酸序列进行比对、蛋白质序列和蛋白质序列进行比对、假设翻译后核酸序列于蛋白质序列进行比对,选择相应的子程序: blastn 是用于核酸对核酸 blastp 是蛋白质对蛋白质序列等等,一共5个自程序。 -i: i 是input的简写,意思是输入文件,就是你自己的要进行比对的序列文件(fasta格式) -d: d是database的简写,意思是要比对的目标数据库,在例子中就是humanRNA.fasta (别忘了要formatdb) -o: o是output的简写,意思是结果文件名字,这个根据你自己的习惯起名字,可以带路径,(上边两个参数-i -d 也都可以带路径) *注意以上4个参数是必须的,缺一不可,下面的参数是为了得到更好的结果自己可调的参数,如果你不加也没有关系,blastall程序本身会给一个默认值! -a: 是指计算时要用的CPU个数,我的机器有两个CPU,所以用-a 2,这样可以并行化进行计算,提高速度,当然你的计算机就一个CPU,可以不用这个参数,系统默认值为1,就是一个CPU -F: 是filter的简写,blastall程序中有对简单的重复序列和低复杂度的一些repeats过滤调,默认是T (注意以后的有几种参数就两个选项,T/F T就是ture,真,你可以理解为打开该功能; F就是false,假,理解为关闭该功能) -T: 是HTML的简写,是指blast结果文件是否用HTML格式,默认是F!如果你想用IE看,我建议用-T T -e: 是Expectation value,期望值,默认是10,我用的10-10! BLASTALL 用法 a.格式化序列数据库 格式化序列数据库— —formatdb formatdb简单介绍: formatdb处理的都是格式为 ASN.1和FASTA,而且不论是核苷酸序列数据库,还是蛋白质序列数据库;不论是使用Blastall ,还是Blastpgp,Mega Blast应用程序,这一步都是不可少的。 formatdb命令行参数: formatdb - 得到formatdb 所有的参数显示(见附录二)和介绍, 主要参数的说明:
本地Blast使用说明 一、软件的下载安装 1.1安装流程 建议安装在非系统盘,如将下载的 BLAST 程序安装到 E:\blast,生成bin、doc 两个子目录,其中 bin 是程序目录,doc 是文档目录,这样就安装完毕了。 1.2 设置环境变量 右键点击“我的电脑”-“属性”,然后选择“高级系统设置”标签-“环境变量”(图1),在用户变量下方“Path”随安装过程已自动添加其变量值,即“E:\Blast\bin”。此时点击“新建”-变量名“BLASTDB”,变量值为“E:\Blast\db”(即数据库路径,图2)。 二、查看程序版本信息 点击 Windows 的“开始”菜单下的“运行”,输入“cmd”调出 MS-DOS 命令行,转到 Blast 安装目录,输入命令“blastn -version”即可查看版本,若能显示说明本地blast 已经安装成功。 三、使用 3.1本地数据库的构建 下载所需的数据(Fasta格式),将X 放到E:\blast\db 文件夹下,然后调出MS-DOS 命令行,转到E:\blast\db 文件夹下运行以下命令:格式化
数据库,命令为: makeblastdb -in 数据库文件 -dbtype 序列类型(核酸:nul;蛋白:prot)-title database_title-parse_seqids -out database_name-logfile File_Name 格式化数据库后,创建三个主要的文件——库索引(indices),序列(sequences)和头(headers)文件。生成的文件的扩展名分别是:.pin、.psq、.phr(对蛋白质序列)或.nin、.nsq、.nhr(对核酸序列)。而其他的序列识别符和索引则包含在.psi和.psd(或.nsi 和.nsd)中。 3.2核酸序列相似性搜索 blastn -db database_name -query input_file -out output_file -outfmt "7 qacc sacc qstart qend sstart send length bitscore evalue pident ppos" 备注:qacc:查询序列Acession号;sacc:目标序列Acession号; qstart qend:分别表示查询序列比对上的起始、终止位置; sstart send:分别表示目标序列比对上的起始、终止位置; length:长度; bitscore:得分; evalue:E-Value值; pident:一致性; ppos:相似性 3.3 查看并获取目标序列: blastdbcmd -db refseq_rna -entry 224071016 -out test.fa 可以从数据库中提取gi号为224071016的序列,并且以fasta格式存入文 件 3.4蛋白质序列相似性搜索 Blastp -db database_name-query input_file -out output_file -outfmt "7 qacc sacc qstart qend sstart send length bitscore evalue pident ppos" 3.5 查看并获取目标序列:重复3.3
Blast本地化:window平台下blast软件的安装boyun发表于 2009-07-09 17:08 | 阅读 1 views 1.对于windows 2000/xp 用户,下载blast- 2.2.18-ia32-win32.exe安装文件 ftp://https://www.doczj.com/doc/5f1809609.html,/blast/executables/LATEST/blast- 2.2.18-ia32-win32.exe 2.创建一个新目录,例如C:\blast,将下载的文件blast-2.2.18-ia32-win32.exe复制到该目录,双击这个文件,自解压产生bin、data、doc 三个目录,bin是程序目录,data是程序使用数据的目录,doc是文档目录。 表:bin目录中的程序 程序说明 bl2seq.exe进行两条序列比对 blastall.exe做普通的blast比对 blastclust.exe blastpgp.exe copymat.exe fastacmd.exe通过gi号,接收号等,在数据库中检索序 列 formatdb.exe格式化数据库 formatrpsdb.exe impala.exe makemat.exe megablast.exe megablast程序 rpsblast.exe seedtop.exe 3.用文本编辑器创建一个ncbi.ini文件,文件包含下面内容:[NCBI] Data="C:\blast\data\" 将ncbi.ini文件存放到系统的Windows 或者 WINNT目录。 4.将”C:\blast\bin”目录添加路径中(该步骤非必须,但会给以后的操作带来方便),方法:
NCBI中Blast种类及使用简介 NCBI中Blast种类简介 1. Blast Assembled Genomes 在一个选择的物种基因组序列中去搜索。 2.Basic Blast 2.1 nucleotide blast--- 用核酸序列到核酸数据库中进行搜索,包括3个程序 2.1.1 Blastn----核酸序列(n)到核酸序列数据库中搜索,是一种标准的搜索。 2.1.2 megablast----该程序使用“模糊算法”加快了比较速度,可以用于快速比较两大系列序列。可以用来搜索一匹ESTs序列和大的cDNA或基因组序列, 适用于由于测序或者其他原因形成的轻微的差别的序列之间的比较 2.1.3 discontiguous megablast----与megablast不同的是主要用来比较来自不同物种之间的相似性较低的分歧序列。 2.2 Protein Blast 2.2.1 Blastp ---蛋白质序列到蛋白质序列数据库中搜索,是一种标准的搜索。 2.2.2 psi-blast---位点特异迭代BLAST —用蛋白查询来搜索蛋白资料库的一个程式。所有被BLAST发现的统计有效的对齐被总和起来形成一个多次对齐,从这个对齐,一个位置特异的分值矩阵建立起来。这个矩阵被用来搜索资料库,以找到额外的显著对齐,这个过程可能被反复迭代一直到没有新的对齐可以被发现。 2.2.3 PHI-BLAST---以常规的表达模型为特别位置进行PSI - BLAST检索,找出和待查询序列具有一样的表达模型且具有同源性的蛋白质序列。 2.3 Translating BLAST 2.3.1 blastx----先将待查询的核酸序列按6 种读框翻译成蛋白质序列,然后将翻译出的蛋白质序列与NCBI 蛋白质序列数据库比较。 2.3.2 tblastn-----先将核酸序列数据库中的核酸序列按6 种读框翻译成
NCBI在线BLAST使用方法与结果详解 BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA 数据库中进行相似性比较的分析工具。BLAST程序能迅速与公开数据库进行相似性序列比较。BLAST结果中的得分是对一种对相似性的统计说明。 BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。 Blast中常用的程序介绍: 1、BLASTP是蛋白序列到蛋白库中的一种查询。库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。 2、BLASTX是核酸序列到蛋白库中的一种查询。先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。 3、BLASTN是核酸序列到核酸库中的一种查询。库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。 4、TBLASTN是蛋白序列到核酸库中的一种查询。与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。 5、TBLASTX是核酸序列到核酸库中的一种查询。此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。 下面是具体操作方法 1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。不同的blast程序上面已经有了介绍。这里以常用的核酸库作为例子。
2,粘贴fasta格式的序列。选择一个要比对的数据库。关于数据库的说明请看NCBI在线blast数据库的简要说明。一般的话参数默认。 3,blast参数的设置。注意显示的最大的结果数跟E值,E值是比较重要的。筛选的标准。最后会说明一下。
新版blast本地化构建+数据库下载+序列间的相似性检索 Ethnobotany 前面记录了blast-2.2.23-ia32-win32的本地化构建及相似性检索,NCBI新近对blast程序做了一些修改推出了blast+,这里结合网上资料、blast+的user manual对blast+的本地化构建及使用作一引荐。 1blast+的本地化构建 链接到:ftp://https://www.doczj.com/doc/5f1809609.html,/blast/executables/blast+/LATEST 下载ncbi-blast-2.2.23+-ia32-win32.tar.gz(绿色版),解压到d盘,并将文件夹更名为blast(我习惯这样做,因为在dos中写命令时方便),这样就安装完毕了,blast下具2个文件即bin 和doc。 2 数据库下载 2.1法1:直接从NCBI下载subject序列去掉txt的扩展名做成数据库即*db,然后将query 序列的txt扩展名掉做成查询文件*in。(格式必须是fasta,名字可以自己随便命名) 2.2法2:从NCBI中的ftp库下载所需要的某一个库或几个库,其链接为 ftp://https://www.doczj.com/doc/5f1809609.html,/blast/db/ 2.3法3:利用新版blast自带的update_blastdb.pl进行下载,这需要安装perl程序。 2.3.1 perl程序的下载和安装 可google“Perl for Windows”获得,也可直接按此连接 https://www.doczj.com/doc/5f1809609.html,/releases.html下载并,安装到任何盘均可。 2.3.2运行update_blastdb.pl进行下载 2.3.2.1开始>运行>cmd+确认>进入dos系统>输入以下命令打开bin文件夹。 2.3.2.2接着输入下述命令回车查看操作帮助(这一步可以不做,不妨碍后续操作) 2.3.2.3还可输入下述命令回车查看NCBI中的库(无需登录NCBI你就可以看到你所需要的库) 2.3.2.4以下载载体库(vector)为例演示如何下载库。输入如下命令回车即可。 直到后面出现done即表示已经下载完毕。如果下载其他数据库,你就可以在上面的perl update_blastdb.pl 后面的vector换成其它数据库的名字即可。 再做本地blast时即可以你下载的压缩文件名代替你bin中*db数据库,进行搜索。 上述三种方法各有优缺点,前两种下载速度较快,但是每次进行检索都需要对数据库进
N C B I在线B L A S T使用 方法与结果详解 This model paper was revised by the Standardization Office on December 10, 2020
N C B I在线B L A S T使用方法与结果详解 BLAST(Basic Local Alignment Search Tool)是一套在蛋白质数据库或DNA数据库中进行相似性比较的分析工具。BLAST程序能迅速与公开数据库进行相似性序列比较。BLAST结果中的得分是对一种对相似性的统计说明。 BLAST 采用一种局部的算法获得两个序列中具有相似性的序列。 Blast中常用的程序介绍: 1、BLASTP是蛋白序列到蛋白库中的一种查询。库中存在的每条已知序列将逐一地同每条所查序列作一对一的序列比对。 2、BLASTX是核酸序列到蛋白库中的一种查询。先将核酸序列翻译成蛋白序列(一条核酸序列会被翻译成可能的六条蛋白),再对每一条作一对一的蛋白序列比对。 3、BLASTN是核酸序列到核酸库中的一种查询。库中存在的每条已知序列都将同所查序列作一对一地核酸序列比对。 4、TBLASTN是蛋白序列到核酸库中的一种查询。与BLASTX相反,它是将库中的核酸序列翻译成蛋白序列,再同所查序列作蛋白与蛋白的比对。 5、TBLASTX是核酸序列到核酸库中的一种查询。此种查询将库中的核酸序列和所查的核酸序列都翻译成蛋白(每条核酸序列会产生6条可能的蛋白序列),这样每次比对会产生36种比对阵列。 NCBI的在线BLAST: 下面是具体操作方法 1,进入在线BLAST界面,可以选择blast特定的物种(如人,小鼠,水稻等),也可以选择blast所有的核酸或蛋白序列。不同的blast程序上面已经有了介绍。这里以常用的核酸库作为例子。 2,粘贴fasta格式的序列。选择一个要比对的数据库。关于数据库的说明请看NCBI在线blast数据库的简要说明。一般的话参数默认。 3,blast参数的设置。注意显示的最大的结果数跟E值,E值是比较重要的。筛选的标准。最后会说明一下。 4,注意一下你输入的序列长度。注意一下比对的数据库的说明。 5,blast结果的图形显示。没啥好说的。 6,blast结果的描述区域。注意分值与E值。分值越大越靠前了,E值越小也是这样。7,blast结果的详细比对结果。注意比对到的序列长度。评价一个blast结果的标准主要有三项,E值(Expect),一致性(Identities),缺失或插入(Gaps)。加上长度的话,就有四个标准了。如图中显示,比对到的序列长度为1405,看Identities这一值,才匹配到1344bp,而输入的序列长度也是为1344bp(看上面的图),就说明比对到的序
BLAST 2.2.28详解本人亲测 1程序下载 链接到:ftp://https://www.doczj.com/doc/5f1809609.html,/blast/executables/blast+/2.2.28/ 下载最新的BLAST+ 程序包,推荐版本ncbi-blast-2.2.28+-win32.exe,版本:ncbi-blast-2.2.28+-win32.exe适用于windows32 位系统,ncbi-blast-2.2.28+-win64.exe 适用Windows 64位系统,请注意选择。 2安装流程 建议安装在非系统盘,如将下载的BLAST 程序安装到D:\blast-2.2.28+ ,生成bin、doc两个子目录,其中bin是程序目录,doc是文档目录,这样就安装完毕了。如图1 图1 程序安装位置 3用户环境变量设置 右键点击“我的电脑”-“属性”,然后选择“高级系统设置”标签-“环境变量”(图1),在用户变量下方“Path”随安装过程已自动添加其变量值,即“D:\blast-2.2.28+\bin”。此时点击“新建”-变量名“BLASTDB”,变量值为“D:\blast-2.2.28+\db”(即数据库路径)。如图2
图2 用户环境变量设置 4查看程序版本信息 点击Windows的“开始”菜单,点击“运行”,输入“cmd”(图3-1,图3-2),调出MS-DOS命令行,在命令行中输入“D:”,输入后按下enter键就进入D盘中,输入cd blast-2.2.28+,转到D:\blast-2.2.28+\。安装目录,输入命令“blastn-version”即可查看版本(图3-3): 图3查看程序版本信息 看到图3 显示说明本地blast已经安装成功。 5blast-2.2.28+本地数据库的构建 数据的获取 法1 :直接从NCBI 或者其他数据库网站下载所需序列做成数据库,或者自己已有的测序数据(格式必须是fasta,名字可以自己随便命名,具体做法下面有说明)。 法2 :从NCBI中的ftp库下载所需要的某一个库或几个库,其链接为ftp://https://www.doczj.com/doc/5f1809609.html,/blast/db/FASTA/其中nr.gz为非冗余的数据库,nt.gz为核酸数据库,month.nt.gz为最近一个月的核酸序列数据。下载的month.nt.gz先用winrar 解压缩,然后用makeblastdb.exe格式化。 法3 :利用新版blast自带的update_blastdb.pl进行下载,这需要安装perl
64位LINUX下BLAST+的本地化 以我的计算机(用户名为yonpen)和数据库nr为例,运行psiblast得到PSSM矩阵。 2012-02-18 1下载程序 在ftp://https://www.doczj.com/doc/5f1809609.html,/blast/executables/blast+/LATEST/下载 ncbi-blast-2.2.25+-x64-linux.tar.gz 2 解压 如解压到用户的主目录(/home/yonpen)下,把解压后的文件夹重新命名为blast,则BLAST+的所有程序在目录/home/yonpen/blast/bin下。 3 添加环境变量 打开终端(Terminal),切换为root用户,执行vim /etc/profile 在最末尾添加export PATH=”/home/yonpen/blast/bin:$PATH”,保存退出。 或直接找到/etc/profile这个文件,在最末尾添加export PATH=”/home/yonpen/blast/bin:$PA TH” 此处若成功,则执行blastn -version会出现版本信息。 4 新建 在目录/home/yonpen/blast下新建一个文件夹,命名为db 在/home/yonpen下新建一个文件,命名为.ncbirc 在文件中添加内容 [BLAST] BLASTDB=/home/yonpen/blast/db 5 下载FASTA格式的数据库 ftp://https://www.doczj.com/doc/5f1809609.html,/blast/db/FASTA/ 如下载nr.gz 6 建立BLAST+可用的数据库 打开终端(Terminal),切换到/home/yonpen/blast/db目录下,执行: makeblastdb –in nr -parse_seqids -hash_index -dbtypeprot 7 使用程序 如使用psiblast 在目录/home/yonpen/blast下新建3个文件夹,分别命名为pssm,input,output 设待查询序列所在文件的名字为3.fasta(一个文件放一条序列,且必须为fasta格式) 执行命令: psiblast -comp_based_stats 1 -evalue 0.001 -num_iterations 3 -db nr -query input/3.fasta -out output/3.txt -out_ascii_pssmpssm/3.pssm
Windows平台下新版blast(2.2.24+)本地化构建+数据库下载+序列间的相似性检索从blast-2.2.23-ia32-win32这个版本开始,本地化blast的参数有了很大改变,NCBI新近对blast 程序做了一些修改推出了blast+,目前最新版本为ncbi-blast-2.2.24+-ia32-win32。与之前的blast相比,新的blast+将blastn,blastx 等合作与blastall命令分隔开来,对各个命令的参数定制更加方便而网上相关的一些教程大同小异,一部分操作已经不适用了,遂整理如下,仅供参考,不当之处,敬请指正。 blast+的本地化构建 1.1程序下载链接到:ftp://https://www.doczj.com/doc/5f1809609.html,/blast/executables/blast+/LATEST 下载最新的BLAST+程序包,推荐版本ncbi-blast-2.2.24+-ia32-win32.tar.gz(绿色版windows32位系统),其他版本:ncbi-blast-2.2.24+-win32.exe适用于windows32位系统,ncbi-blast-2.2.24+-win64.exe 适用Windows 64 位系统,请注意选择。 1.2安装流程 建议安装在非系统盘,如将下载的BLAST 程序安装到E:\blast,生成bin、doc 两个子目录,其中bin 是程序目录,doc是文档目录,这样就安装完毕了。 1.3用户环境变量设置 右键点击“我的电脑”-“属性”,然后选择“高级系统设置”标签-“环境变量”(图1),在用户变量下方“Path”随安装过程已自动添加其变量值,即“E:\Blast\bin”。此时点击“新建”-变量名“BLASTDB”,变量值为“E:\Blast\db”(即数据库路径,图2)。
Blast软件的详细使用方法 blastall -p blastn -i myRNA.fasta -d humanRNA.fasta -o myresult.blastout -a 2 -F F -T T -e 1e-10 解释如下: blastall: 这是本地化/命令行执行blast时的程序名字!(Tips:blastall直接回车就会给出你所有的参数帮助,但是英文的) -p: p 是program的简写,program在计算机领域中是程序的意思。此参数是指定要使用何种子程序,所谓子程序,就是针对不同的需要,如核酸序列和核酸序列进行比对、蛋白质序列和蛋白质序列进行比对、假设翻译后核酸序列于蛋白质序列进行比对,选择相应的子程序: blastn 是用于核酸对核酸blastp 是蛋白质对蛋白质序列等等,一共5个自程序。 -i: i 是input的简写,意思是输入文件,就是你自己的要进行比对的序列文件(fasta格式)-d: d是database的简写,意思是要比对的目标数据库,在例子中就是humanRNA.fasta (别忘了要formatdb) -o: o是output的简写,意思是结果文件名字,这个根据你自己的习惯起名字,可以带路径,(上边两个参数-i -d 也都可以带路径) *注意以上4个参数是必须的,缺一不可,下面的参数是为了得到更好的结果自己可调的参数,如果你不加也没有关系,blastall程序本身会给一个默认值! -a: 是指计算时要用的CPU个数,我的机器有两个CPU,所以用-a 2,这样可以并行化进行计算,提高速度,当然你的计算机就一个CPU,可以不用这个参数,系统默认值为1,就是一个CPU -F: 是filter的简写,blastall程序中有对简单的重复序列和低复杂度的一些repeats过滤调,默认是T (注意以后的有几种参数就两个选项,T/F T就是ture,真,你可以理解为打开该功能; F就是false,假,理解为关闭该功能) -T: 是HTML的简写,是指blast结果文件是否用HTML格式,默认是F!如果你想用IE看,我建议用-T T -e: 是Expectation value,期望值,默认是10,我用的10-10! BLASTALL 用法 a.格式化序列数据库 格式化序列数据库——formatdb formatdb简单介绍: formatdb处理的都是格式为ASN.1和FASTA,而且不论是核苷酸序列数据库,还是蛋白质序列数据库;不论是使用Blastall ,还是Blastpgp,Mega Blast应用程序,这一步都是不可少的。 formatdb命令行参数: formatdb - 得到formatdb 所有的参数显示(见附录二)和介绍, 主要参数的说明: -i 输入需要格式化的源数据库名称Optional -p 文件类型,是核苷酸序列数据库,还是蛋白质序列数据库 T – protein F - nucleotide [T/F] Optional default = T -a 输入数据库的格式是ASN.1(否则是FASTA) T - True, F - False. [T/F] Optional default = F
Windows系统下本地BLAST安装方法 1.下载安装文件: 以blast-2.2.23-ia32-win32.exe为例,将此安装文件放至指定目录,以G:\blast-\为例,如图所示: 2. 运行安装程序: 双击上述安装文件,单击运行: 程序会自动在blast-文件夹下生成3个文件夹:\bin\、\data\和\doc\:
3. 添加配置文件: 在桌面(任意可以新建文件的地方)新建一个.txt文件,然后将其重命名为NCBI.ini,在提示更改后缀名的对话框中点是。打开NCBI.ini,在其中写入如下两行内容: [NCBI] Data="path\data\" 上边的path是你的blast安装路径,在本例中为G:\blast-,因此,NCBI.ini中的内容为: [NCBI] Data="G:\blast-\data\" 写完后保存,然后将该文件复制至C:\Windows目录下: 至此,本地blast-2.2.23-ia32-win32安装完毕。 4. 导入数据库:
从ftp://https://www.doczj.com/doc/5f1809609.html,/blast/db/上,可下载各类数据库文件,下载完毕后,将其解压至G:\blast-\data\目录下。 注意事项: 1.NCBI.ini中的路径为blast所在安装路径; 2.此安装办法适用与指定版本,对于blast+版本不适用,若想安装新 版本,可自行到网站查阅安装办法; 附:运行示例: 1.打开cmd命令行;
2.通过cd命令到达安装目录的bin\目录下 3.通过dir命令查看全部可执行的子程序: 4.使用blastall.exe进行比对: 输入blastall.exe -d refseq_rna.01 -i G:\blast-\data\test_query.fa -p blastn 该命令各部分的含义为: ①blastall.exe:blast主程序; ②-d refseq_rna.01:选择refseq_rna.01为被搜索的数据库,其数
本地blast的安装及使用 安装: 1.首先进入NCBI 2.点击ALL Resources 3.点击ALL Resources里的Downloads选项卡 4.点击BLAST(Stand-alone)选项 在BLAST+executables中点击 ftp://https://www.doczj.com/doc/5f1809609.html,/blast/executables/blast+/LATEST/ . 链接(这只是说这个链接如何找到的,可以直接点击这个链接进行下载)。 5点击ncbi-blast-2.2.29+-win32.exe进行下载,大家的电脑一般为32位的, 加入为64 位的则需要点击ncbi-blast-2.2.29+-win64.exe下载,根据个人情况定 6下载好后点击“下一步“进行安装。 运行: 1.点击电脑桌面的“开始“——”运行“,在”打开“中输入” cmd“,(这也就是调取DOS命令,快捷键”windows“+“R“键,然后回车) 2切换到blast的bin目录下,例如我的路径是C:\Program Files\NCBI\blast-2.2.29+\bin,那么我的命令是: 然后回车。
切换后的结果是: 3把你的物种数据和比对的数据文件移动到bin文件夹下,然后做下面的。 1)建库根据你要比对的物种序列建库 dos 命令:makeblastdb -in ~ -dbtype nucl/prot -out ~ in 后面的‘~’里填要建库的序列文件名称,如整个水稻蛋白质组 第二个‘~’里填库的名称(自己命名) nucl :建核苷酸库,prot:建蛋白质库(根据你数据要求任选一个) 2)比对 dos 命令:blastp/blastn -query ~ -db ~ -out ~ -evalue ~ -outfmt blastp 为比对蛋白质序列,blastn比对核苷酸序列 query后面的‘~’填你要比对的序列文件名 db 后面填你第一步建好库的名称 out 输出最终结果名称 evalue 你自己设一个期望值(5) outfmt 输出文件格式填数字6或7 (1)建库 结果 (2)比对:
Alignment: 序列比对。将两个或多个序列排在一起,以达到最大一致性的过程(对于氨基酸序列是比较它们的保守性),这样可以评估序列间的相似性和同源性。Algorithm: 算法。在计算机程序中包含的一种固定过程。 Bit score: 二进制。二进制值S'源于统计性质被数量化的打分系统中产生的原始比对分数S。由于二进制值相对于打分系统已经被标准化,它们常用于比较不同搜索之间的比对分数。 BLOSUM: 模块替换矩阵。在替换矩阵中,每个位置的打分是在相关蛋白局部比对模块中观察到的替换的频率而获得的。每个矩阵被修改成一个特殊的进化距离。例如,在BLOSUM62矩阵中,是使用一致性不超过62%的序列进行配对来获得打分值的。一致性大于62%的序列在配对时用单个序列表示,以避免过于强调密切相关的家族成员。Conservation: 保守。指氨基酸或DNA(普遍性较小)序列某个特殊位置上的改变,并不影响原始序列的物理化学性质。 Domain: 结构域。蛋白质在折叠时与其他部分相独立的一个不连续的部分,它有着自己独特的功能。 DUST: 一个低复杂性区段过滤程序。 E value: E值。期望值。在一个数据库中所搜索到的打分值等于或大于S的不同比对的个数。E值越低,表明该打分值的显著性越好。 Filtering: 过滤,也叫掩蔽(masking)。指对那么经常产生乱真的高分数的核苷酸或氨基酸序列区域进行隐藏的过程。 Gap: 空位。在两条序列比对过程中需要在检测序列或目标序列中引入空位,以表示插入或删除。为了避免在比对时出现太多的空位,可以在收入空位的同时,从比对的打分值中减去一个固定值(空位值)。在多余的核苷酸或氨基酸周围引入空位时,也要对比对的打分值进行罚分。 Global Alignment: 整体联配。对两个核苷酸或蛋白质序列的全长进行的比对。 H: 相对熵值。目标残基和底物残基频率的相对熵记作H。H可以衡量某个位置(这个位置可以通过概率来区分比对)上由于偶然因素而得到的平均信息(用字节表示)。H值越高,短的比对就越可以通过概率来区分;H值越低,需要的比对长度越长。 Homology: 同源性。由共同的祖先所遗传得到的相似性。 HSP: High-scoring segment pair,高打分值片段。在一个给定的搜索中,没有空位的局部比对能得到最高的比对打分值。 Identity: 一致性。两个(核苷酸或氨基酸)序列比对时不变部分的长度。 K: K值。用来计算BLAST程序中打分函数的一个统计参数。它可以看作搜索空间大小的一个自然衡量尺度。K值通常用于将原始比对值S转换为二进制值S'。 Lambda: λ值。用来计算BLAST程序中打分函数的一个统计参数;它可以看作打分系统的一个自然衡量尺度。λ值通常用于将原始比对值S转换为二进制值S'。 Local Alignment: 局部联配。对两个核苷酸或蛋白质序列的一部分所进行的比对。 Low Complexity Region(LCR): 低复杂性区域。指组分(包括均聚物、短周期重复片段)区域和有许多单个或多个残基的区域。SEG程序用来筛选或过滤氨基酸序列中低复杂性区域。DUST程序用来筛选或过滤核苷酸序列中的低复杂性区域。 Masking: 掩蔽。也叫过滤(filtering),指为了提高对序列相似性搜索是时的敏感性,而从序列中移除重复的或低复杂性区域的过程。
用Perl 实现在W indows 下本地化运行BLAST 范彦辉,陶士珩 (西北农林科技大学生物信息学研究中心,陕西杨凌712100) 摘要:通过编写Perl 脚本实现BLAST 的本地化运行,使不懂编程的人也能在windows 下进行自己的BLAST 。关键词:BLAST Perl 序列比对 中图分类号:TP316 文献标识码:A 文章编号:1672-5565(2008)-04-178-04 收稿日期:2007-10-19;修回日期:2008-01-08 作者简介:范彦辉(19-),男,河南信阳,硕士研究生,生物信息学,yanhuifan @https://www.doczj.com/doc/5f1809609.html, 3通讯作者:陶士珩(19-),男,教授,博士生导师,生物信息学,shihengt @https://www.doczj.com/doc/5f1809609.html, Run stand -alone B LAST on Windows using Perl script FAN Y an -hui ,T AO Shi -heng 3 (Bioinformatics center ,Northwest A &F Univer sity ,Yangling Shanxi 712100,China ) Abstract :T o run stand -alone BLAST on windows operating system ,a Perl script was written to help those who know little about programming language. K ey Words :BLAST Perl Sequence alignment 1 软件安装 1.1 Perl 在windows 操作系统下的安装 Perl 是Practical Extraction Report Language (实用析取报表 语言)的缩写,他是由Larry Wall 最初设计编写的。Perl 是被设计成跨平台运行的,可用于UNIX 、Linux 、M AC 和Windows 等环境下编程[1]。Perl 5的Windows 版可以从Internet 上免费下载。ActivePerl 是由ActiveS tate 公司(w w w.ActiveS https://www.doczj.com/doc/5f1809609.html, )开发和维护的,是完全Win 32化的Perl ,对于支持Windows 编程较好。ActivePerl 目前用于Windows 操作系统的最新版本为ActivePerl -5.8.8.822-MSWin32-x86-280952,下载地址为http ://https://www.doczj.com/doc/5f1809609.html, ΠActivePerl ΠWindows Π5.8Π ActivePerl -5.8.8.822-MSWin32-x86-280952.msi 。双击安 装文件,按照默认设置安装,将被安装在C 盘根目录下。位置为C :\Perl ,包含bin 、eg 、etc 、html 、lib 和site 六个文件夹。 1.2 BLAST 在windows 操作系统下的安装 BLAST 是Basic Local Alignment Search T ool (基本局部比对 搜索工具)的缩写 [2] 。BLAST 安装程序可以到NC BI (http :ΠΠ w w w.ncbi.nlm.nih.g ov Π )下载。目前用于Windows 操作系统的最新版本为blast -2.2.17-ia32-win32,下载地址为ftp :ΠΠ ftp.ncbi.nih.g ov Πblast Πexecutables Πrelease Π2.2.17Πblast -2.2.17-ia32-win32.exe 。双击安装文件,生成bin 、data 和doc 三个 文件夹。将这三个文件拷贝到C :\blast 目录下(使用者可自由选择目录,下文均使用此目录)。然后在C :\WI NDOWS 下创建一个名为NC BI.ini 的配置文件,用记事本打开,写入以下两行代码 [3] : [NC BI] Data =“path Πdata Π” path Πdata 是指blast 程序中data 所在的地址,在本文中,C :\WI NDOWS \NC BI.ini 文件的内容是:[NC BI] Data =C :Πblast Πdata 2 BLAST 的本地化运行 2.1 通过“命令提示符”实现本地化运行 运行BLAST 必须有用来查询的序列和被查询的数据库。数据库可以从NC BI 的BLAST 数据库(ftp :ΠΠftp.ncbi.nih. g ov Πblast Πdb Π )下载。S tand -aloneBLAST 的最大优势在于用户可以将自己感兴趣的FAST A 格式的蛋白质或核酸序列制作成BLAST 数据库。本文使用从NC BI 下载的yeast.nt.gz (3645K B ,ftp ://ftp.ncbi.nih.g ov/blast/db/FAST A/yeast.nt.gz ) 作为数据库,该文件解压缩后得到yeast.nt 。创建一个数据库文件夹(C :\blast \database ),将yeast.nt 放到该文件夹内。 第一步:数据库格式化。 启动“命令提示符”窗口(开始->所有程序->附件->命令提示符),进入BLAST 应用程序所在目录(本文使用 命令:cd c :\blast \bin )。使用formatdb 命令对yeast.nt 数据库进行格式化。通过运行命令(formatdb -)可以得到format 2 db 的版本号及运行参数 [4] ,本文所得参数见表1。 对yeast.nt 格式化的命令为: formatdb -i c :\blast \database \test.nt -p f -o t 命令运行完成后,在C :\blast \database 文件夹下会生成一系列文件,这些文件是进行BLAST 所必需的。注意,格式化完成后,原数据库文件并不是进行BLAST 所必须的[4]。 生物信息学 China Journal of Bioinformatics 技术与方法