
中国科学: 生命科学
2012年 第42卷 第5期: 345 ~ 354 SCIENTIA SINICA Vitae
https://www.doczj.com/doc/74718437.html, https://www.doczj.com/doc/74718437.html,
英文引用格式: Sun C, Li J, Zhou B. Mechanism of action of artemisinins: a long unsettled challenge. SCIENTIA SINICA Vitae, 2012, 42: 345–354, doi:
10.1360/052012-168
《中国科学》杂志社
SCIENCE CHINA PRESS
评 述
青蒿素类药物的作用机制: 一个长久未决的基础 研究挑战
孙辰, 李坚, 周兵*
清华大学生命科学学院, 生物膜与膜生物国家重点实验室, 北京 100084 * 联系人, E-mail: zhoubing@https://www.doczj.com/doc/74718437.html, 收稿日期: 2012-04-20; 接受日期: 2012-04-24 doi: 10.1360/052012-168
摘要 青蒿素是中国自主研制的抗疟良药, 高效、低毒, 许多基于青蒿素研发的衍生物具有良好的抗疟效果, 近年来已成为抗疟的一线药物, 受到世界医疗卫生界的充分肯定. 虽然青蒿素结构奇特, 抑疟效果显著, 但40年来其生物作用机制之谜一直未被彻底破解. 针对青蒿素类药物的作用机制, 提出了不同的假说, 如血红素参与青蒿素的激活并被烷基化从而起到抑疟作用, 线粒体参与青蒿素的激活和作用过程, 某些特定的蛋白是青蒿素作用靶点等. 除抑疟外, 青蒿素类药物在杀灭其他种类寄生虫、抑制某些癌症细胞以及抗病毒、治疗类风湿等方面也有一定作用. 本文将对青蒿素类药物作用机制的研究进行综述及展望, 包括抗疟疾过程中的药物激活、作用靶点以及简要的青蒿素抑制肿瘤细胞作用机制, 以期为今后的研究提供帮助.
关键词
青蒿素 作用机制 血红素 线粒体
2011年9月, 中国中医科学院科学家屠呦呦获得美国拉斯克临床医学奖, 以表彰她在抗疟药物青蒿素(artemisinin)开发过程中的贡献. 一时间, 疟疾和抗疟良药青蒿素成为国人关注的焦点. 疟疾是严重影响人体健康的传染性疾病, 尤其在非洲、南亚、东南亚及南美洲大陆的热带亚热带地区发病严重. 世界卫生组织统计报告显示, 疟疾2010年发病人数为2.16亿, 造成约65.5万人死亡, 其中86%的受害者是5岁以下儿童[1].
青蒿素的发现是中国20世纪70年代“中国疟疾防治药物研究工作协作项目”(又称“523”科研项目)中的一项重要成果, 中国科学家们的协作研发为世界人民抗击疟疾做出了重要贡献. 青蒿素对红内期疟
原虫有直接杀灭作用, 快速高效且毒性低, 缺点是半衰期比较短, 单独使用再燃率较高. 其他治疗疟疾的药物, 如氯喹、甲氟喹和奎宁等均出现了抗药株, 而青蒿素类药物自投入使用以来, 除在柬埔寨地区出现青蒿琥酯对疟原虫清除效率降低的病例(体外实验并没有出现类似的延缓现象)[2], 尚无确证的青蒿素类药物抗性疟原虫出现. 为防止将来青蒿素抗性菌株的出现, 世界卫生组织颁布了 “青蒿素联合疗法”(artemisinin combination therapy, ACT).
除治疗疟疾的良好效果外, 青蒿素在杀灭其他种类寄生虫, 抑制某些癌症细胞以及抗病毒、治疗类风湿等方面也有一定作用. 青蒿素的治疗效果举世瞩目, 但其作用机制至今仍是一个谜, 也是一个长期
孙辰等: 青蒿素类药物的作用机制: 一个长久未决的基础研究挑战
346
存在争议的话题. 本文将对青蒿素作用机制的研究现状进行综述.
1 青蒿素简介
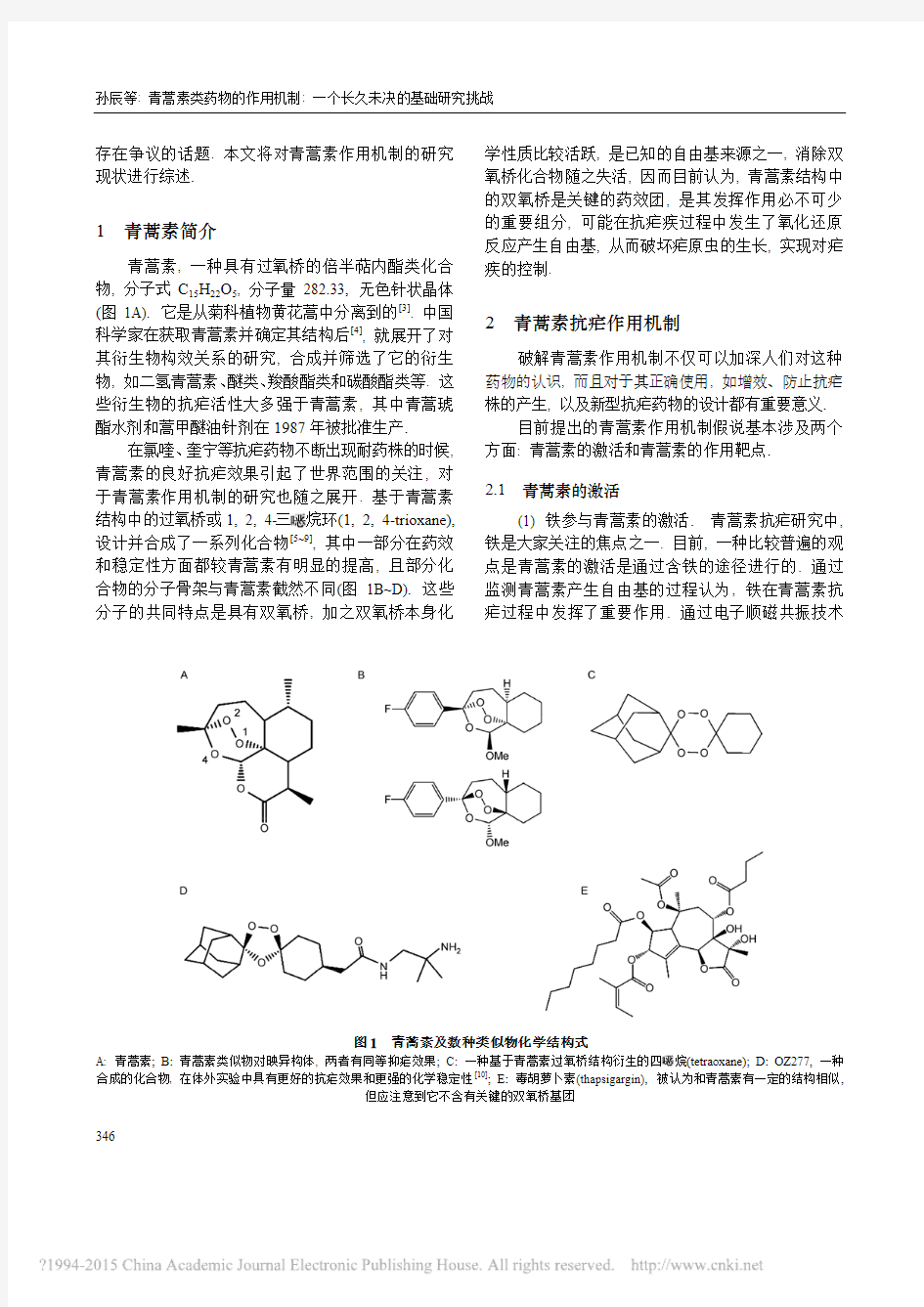
青蒿素, 一种具有过氧桥的倍半萜内酯类化合物, 分子式C 15H 22O 5, 分子量282.33, 无色针状晶体(图1A). 它是从菊科植物黄花蒿中分离到的[3]. 中国科学家在获取青蒿素并确定其结构后[4], 就展开了对其衍生物构效关系的研究, 合成并筛选了它的衍生物, 如二氢青蒿素、醚类、羧酸酯类和碳酸酯类等. 这些衍生物的抗疟活性大多强于青蒿素, 其中青蒿琥酯水剂和蒿甲醚油针剂在1987年被批准生产.
在氯喹、奎宁等抗疟药物不断出现耐药株的时候, 青蒿素的良好抗疟效果引起了世界范围的关注, 对于青蒿素作用机制的研究也随之展开. 基于青蒿素结构中的过氧桥或1, 2, 4-三烷环(1, 2, 4-trioxane), 设计并合成了一系列化合物[5~9], 其中一部分在药效和稳定性方面都较青蒿素有明显的提高, 且部分化合物的分子骨架与青蒿素截然不同(图1B~D). 这些分子的共同特点是具有双氧桥, 加之双氧桥本身化
学性质比较活跃, 是已知的自由基来源之一, 消除双氧桥化合物随之失活, 因而目前认为, 青蒿素结构中的双氧桥是关键的药效团, 是其发挥作用必不可少的重要组分, 可能在抗疟疾过程中发生了氧化还原反应产生自由基, 从而破坏疟原虫的生长, 实现对疟疾的控制.
2 青蒿素抗疟作用机制
破解青蒿素作用机制不仅可以加深人们对这种药物的认识, 而且对于其正确使用, 如增效、防止抗疟株的产生, 以及新型抗疟药物的设计都有重要意义.
目前提出的青蒿素作用机制假说基本涉及两个方面: 青蒿素的激活和青蒿素的作用靶点.
2.1 青蒿素的激活
(1) 铁参与青蒿素的激活. 青蒿素抗疟研究中, 铁是大家关注的焦点之一. 目前, 一种比较普遍的观点是青蒿素的激活是通过含铁的途径进行的. 通过监测青蒿素产生自由基的过程认为, 铁在青蒿素抗疟过程中发挥了重要作用. 通过电子顺磁共振技术
图1 青蒿素及数种类似物化学结构式
A: 青蒿素; B: 青蒿素类似物对映异构体, 两者有同等抑疟效果; C: 一种基于青蒿素过氧桥结构衍生的四烷(tetraoxane); D: OZ277, 一种合成的化合物, 在体外实验中具有更好的抗疟效果和更强的化学稳定性[10]; E: 毒胡萝卜素(thapsigargin), 被认为和青蒿素有一定的结构相似,
但应注意到它不含有关键的双氧桥基团
中国科学: 生命科学 2012年 第42卷 第5期
347
(EPR)及捕获剂DMPO 进一步揭示了二者之间的关系: 青蒿素通过一个依赖于铁的通路产生自由基[11]. 如果在反应体系中加入吡哆醛苯甲酰腙(pyridoxal benzo- ylhydrazone)和去铁酮(1,2-dimethyl-3-hydroxypyrid- 4-one)这两种疏水性铁螯合剂, 青蒿素及其衍生物的抑疟效果将减弱. 去铁敏(desferrioxamine), 另一种铁螯合剂, 也降低了青蒿素的作用[12]. Wu 等人[13]发现, 青蒿素在亚铁离子存在下可以与半胱氨酸作用, 得到胱氨酸及一种新的重排产物. 青蒿素分子中的过氧桥结构是其发挥作用的关键药效团. 在青蒿素激活过程中, 关于如何打开过氧桥有两种假说, 一种是通过铁参与还原, 均裂过氧桥产生氧中心自由基进而重排形成碳中心自由基[14~16], 另一种观点是铁起到类似路易斯酸(lewis acid)的作用, 不对称断裂过氧桥, 分子离子化, 之后再借助铁或者非过氧化的氧进行后续反应[17~20], 认为这样的方式所需能量更低、更稳定, 且有可能在该过程中产生能对细胞造成损害的活性氧自由基.
(2) 血红素(heme)参与青蒿素的激活. 疟原虫在传播繁殖过程中有两个寄主, 一个是人体, 另一个是按蚊. 在人体内为无性繁殖. 其长梭形的孢子在肝细胞内形成裂殖体, 成熟后会涨破肝细胞进入血液, 侵入红细胞中增殖. 它们依靠摄取红细胞内的血红蛋白做为自身氨基酸合成的来源, 该过程会产生对虫体不利的分解产物血红素, 血红素形成三价铁二聚体(hematin), 疟原虫体内存在一种机制可以将其转化成没有细胞毒性的色素颗粒(hemozoin)累积于细胞质内, 即疟色素.
有研究表明, 非血红素的铁[11,21]和血红素中的铁[22]均能与青蒿素作用. 两种观点都有相关实验数据支持: 对于前者, 实验将一种特异螯合非血红素来源的铁离子螯合剂与青蒿素偶联之后与未经修饰的青蒿素一同进行抑疟实验, 发现两者具有拮抗作用[23], 表明非血红素来源的铁对青蒿素有重要作用; 而另一部分学者发现, 从用青蒿素处理的恶性疟原虫菌株中可以分离到血红素(heme)与青蒿素的复合物[24], 还有体外实验发现, 将青蒿素与不同形式的铁, 包括血红素、二价铁离子、脱氧的和氧化的血红蛋白在相同的条件下进行反应, 血红素与青蒿素反应的效率远高于其他含铁分子[12], 表明血红素对于激活青蒿素起关键作用. 疟原虫寄生于血红细胞中, 血红素来源丰富. 目前, 究竟是自由铁还是血红素的铁激活青蒿素还存在争议.
(3) 线粒体参与青蒿素的激活. 模式生物是研究药物分子机制的有力工具, 酿酒酵母(Saccharomyces cerevisiae )作为经典模式生物之一, 被用于青蒿素类药物作用机制的探索. 通常情况下, 酿酒酵母主要依赖糖酵解途径, 在发酵型培养基(如以葡萄糖为碳源)上的生长状况更好, 此类型的生长过程不需要线粒体供能, 而当非发酵型碳源(如以甘油或乙醇作为碳源)出现时, 它们的生长必须依赖于线粒体.
研究发现, 青蒿素可以抑制酿酒酵母菌在非发酵培养基上的生长, 但是基本不能抑制在发酵培养基上的生长. 这种现象暗示了青蒿素与线粒体的紧密联系[25]. 通过筛选得到了NDI1, NDE1的突变株, 发现其具有青蒿素抗性. NDI1和NDE1是编码酿酒酵母线粒体电子传递链中NADH 脱氢酶的基因, 这两个基因在恶性疟原虫中存在同源序列, 而哺乳动物中不存在. 哺乳动物线粒体内与这一结构发挥相似功能的是由多个亚基组成的呼吸链复合物Ⅰ. 实验表明, 当敲除NDE1或NDI1基因时, 可以观察到酵母菌对青蒿素的耐受性增强, 敏感性降低; 过量表达则可增强敏感性. 线粒体呼吸链抑制剂DPI(NADH 脱氢酶抑制剂)的加入可以减缓酵母菌细胞和疟原虫细胞对青蒿素的代谢[26], 减弱疟原虫和酵母菌对青蒿素的敏感性. 基于这一模型, 推测青蒿素的还原激活(获得电子)是通过线粒体的还原物, 且很可能是通过获得电子传递链中的电子完成的. 呼吸链在电子传递过程中有时会发生电子泄漏, 电子被O 2获得进而产生自由基, 而青蒿素恰好又是一种过氧化合物. 另值得一提的是, 线粒体特别是呼吸链有丰富的铁以及血红素.
(4) 其他观点. 对于铁参与激活青蒿素这一理论, 也有部分研究者持不同意见, 如Haynes 等人[27]在寻找新型青蒿素衍生物过程中发现, 有两种青蒿素烷氨基衍生物表现出了比青蒿素更好的抗疟作用, 但是其和铁的作用微弱. 此外, 对其他一些可以介导反应形成过氧化物并作为自由基来源的分子也有研究和报道[19]. 在这些假说中, 异源分解过氧桥先于重排发生, 导致了破坏细胞大分子的羟基自由基的生成. 另有报道也反驳了铁在青蒿素中的作用, 同时提出由二价铁介导的碳自由基的形成不是青蒿素发挥抗疟作用的原因[17].
近来有报道认为, 青蒿素抑制疟疾的活性需要
孙辰等: 青蒿素类药物的作用机制: 一个长久未决的基础研究挑战
348
血红蛋白的消化和吸收, 当抑制了血红蛋白酶的活性, 干扰血红蛋白降解通路或者直接从培养基中去除红细胞裂解产物, 都可以显著减弱青蒿素的抑疟活性[28].
综合上述观点, 有一些问题还存在争议, 例如,若铁确实参与了青蒿素的作用过程, 那么铁在体内的作用是直接的还是间接的, 是以何种形式、在哪一阶段、以何种机制参与反应等. 一种相对被认可的结论是活性氧自由基(ROS)的产生是对细胞造成损伤的重要原因之一. 因此也可以存在这样的假设, 认为可能是青蒿素激活产生少量的ROS, 这些ROS 与铁相互作用, 扰乱了铁平衡, 又激化了更多ROS 的生成, 从而对细胞造成损伤. 有关青蒿素激活的各种观点假说还需更多体内实验进行探究.
2.2 青蒿素类药物的作用靶点
嵌入在青蒿素倍半烯萜内酯骨架中的双氧桥对青蒿素抗疟有着至关重要的作用是进行机制研究的最初线索, 而自由基的生成与双氧桥的断裂有着密切的关系. 早在20世纪80年代末期, 自由基在青蒿素类药物中的作用就得到了实验的证实[29]. 研究发现, 在体外实验中, 自由基清除剂可以拮抗青蒿素类药物的抗疟活性, 同时自由基生成剂能增强其抗疟作用. 相关的体内实验也证实了这一关系[30,31]. 然而, 青蒿素的作用又与通常意义上引起损伤的典型氧化剂不同. 由于青蒿素的一个分子一次反应只能产生一个自由基, 实验观察到的内源性氧化产物只在较高的药物施用浓度下才能被观察到[32,33], 而实际杀灭疟原虫时青蒿素的用量很低, 在nmol/L 级, 所以青蒿素应存在着其特殊的作用方式.
(1) 血红素的烷基化. 不少研究认为, 血红素既是激活青蒿素类药物的裂解还原激活剂, 又是其发挥作用的重要靶标, 青蒿素对血红素的修饰、烷基化是杀灭疟原虫的原因. 早期的血红素模型中青蒿素的作用机制与奎宁相仿, 都是作用于液泡, 通过影响血红素的脱毒来抑制青蒿素. 这一观点有一些相关实验支持, 如当14C 标记的青蒿素加入被红细胞侵染的恶性疟原虫时, 辐射剂量测量显示疟色素中有大量青蒿素. 通过高效液相色谱, 可以在试管中分离血红素-青蒿素复合物[34], 说明两者之间有生理联系. 青蒿素抑制液泡中蛋白水解酶的活性, 体内和体外实验都表明青蒿素可以与血红素结合[35]. 推测认为,
青蒿素的抗疟作用可能是由烷基化血红素产生的疟色素引起的, 也可能是由于形成了一种还原性更强的环境导致的[36]. 在被感染的小鼠中也观察到了烷基化的血红素[37,38]. 体外实验中化学合成血红素-青蒿素复合物能抑制血红素聚合过程, 导致毒性血红素的释放和疟原虫的死亡[39,40]. 一些青蒿素的衍生物虽可以与卟啉产生碳自由基, 但不能发生烷基化反应, 经过检测发现这些衍生物也没有抗疟活性. 研究还发现, 合成的有抑疟活性的1,2,4-三烷衍生物能与卟啉发生反应[41]. 由此认为, 烷基化血红素是发挥抗疟活性的重要因素.
然而, 另一些实验结果和血红素理论有一定冲突. 例如, 红细胞内的疟原虫实验发现, 青蒿素的施用没有减少疟色素的含量[42], 表明青蒿素通过抑制疟色素的形成发挥作用的结论有待修正. 随后研究发现, 10-脱氧二氢青蒿素是一种有效的抗疟药物, 但是并不抑制疟色素的形成[43]. 也有研究发现, 水解稳定、血红素惰性的化合物仍然具有很好的抗疟特性[27]. 此外, Ro40-4388, 一种限制血红素降解第一步反应的蛋白酶抑制剂, 与氯喹有拮抗作用, 与青蒿素既无拮抗作用也没有协同作用, 提示血红素对于青蒿素的抗疟作用不是必需的[12]. 最近, 在CO 环境下进行了青蒿素抗疟作用检测, 实验依据CO 会与血红素结合, 形成羰基血红蛋白(CO-Hb-Fe(Ⅱ))或CO-heme- Fe(Ⅱ), 可能会掩盖青蒿素与血红素的作用. 结果显示, 在CO 环境下, 青蒿素效果好, 而血红素惰性的青蒿素衍生物效果下降, 说明Hb-Fe(Ⅱ)或heme-Fe(Ⅱ)没有参与青蒿素抗疟作用[44]. 因而对于血红素理论的修正观点认为, 血红素只是一种激活剂, 不是重要的靶标[45], 除了激活以外, 血红素可能与青蒿素的降解有关而不是增强它的功能.
(2) 蛋白靶点理论. 青蒿素的一个鲜明特点就是对疟原虫有强烈的特异性而对人不产生副作用, 这使得人们推测青蒿素可能会在疟原虫体内特异激活, 或者疟原虫体内存在着对青蒿素的特异性靶标. 对于大部分药物来说, 都存在着较为专一和特异的靶标.
(ⅰ) PfATP6靶点理论. 这一靶点理论曾风靡一时, 源自于2003年Nature 上的一篇文章, 提出青蒿素特异地抑制恶性疟原虫的肌浆内质网钙ATP 酶(PfATP6)[12]. 作者认为, 青蒿素与毒胡萝卜素在结构上有一定相似性(图1), 而后者能特异性结合并抑制
中国科学: 生命科学 2012年 第42卷 第5期
349
肌浆内质网钙ATP 酶(SERCA). 为了探究青蒿素与疟原虫SERCA 间的关系, 以及在扼杀疟原虫过程中发挥的作用, 将PfATP6在非洲爪蟾卵母细胞中进行了表达. 结果发现, 青蒿素对PfATP6具有强大且特异的抑制效果. 因此, 提出青蒿素结合PfATP6发挥功能的理论. 后续研究发现, 当PfATP6 263位亮氨酸和对应的哺乳动物SERCA 255位的谷氨酸进行互换, 它们的抑制效果也随之改变[46]. 一时间, PfATP6理论备受关注.
但是, 近年来一些与此相仿的嵌合实验, 针对一系列抗疟药, 包括青蒿素及其衍生物、奎宁类药物和其他一些过氧化物与毒胡萝卜素结合, 实验结果没有发现上述关系[47]. 值得注意的是, 某些原来用于支持这一学说的实验不能被重复[48,49]. 青蒿素结构中的过氧桥对抗疟作用的发挥有重要意义, PfATP6模型认为, 青蒿素与毒胡萝卜素作用相似, 都是作用在疟原虫的肌浆内质网钙ATP 酶上并最终导致疟原虫死亡. 但是, 毒胡萝卜素并没有过氧桥结构. 另外, 构效研究结果表明, 许多结构不相同的衍生物都展现出了抗疟活性, 但是对于PfATP6的结合似乎更依赖于分子骨架而非过氧桥, 对于结构千差万别的各种衍生物分子而言, 如何统一青蒿素类药物的抗疟机制非常困难. 最近, 青蒿素与毒胡萝卜素之间的结构相似性也受到了质疑, 认为这两种分子结构相似度极有限[50]. 有研究报道, 在疟原虫体内将野生型PfATP6基因分别替换成含有L263E, S769N 的突变型, 用青蒿素及衍生物处理后并未观察到明显的IC50变化[51,52], 这对PfATP6靶点理论构成强烈的冲击. 在酵母菌中异源表达PfATP6并进行纯化后, 发现其与毒胡萝卜素的亲和性较低, 而与环匹阿尼酸(cyclopiazonic acid)的亲和性较高, 同时也未观察到青蒿素对它的抑制作用. 其他类似的研究也支持这一结果[47,53]. 总之, 这个理论不再被看好, 人们认为, PfATP6即使可以作为其他药物的靶标, 也不太可能是青蒿素的作用靶点[54].
(ⅱ) 其他蛋白靶点理论. 除了PfA TP6靶点理论, 还有研究提出了其他一些蛋白, 被认为可能是青蒿素的作用靶标, 但目前证据较少, 说服力比较薄弱. 其中包括转译控制肿瘤蛋白(TCTP)、消化泡上的膜转运蛋白PfMDR1、PfCRT 、半胱氨酸蛋白酶家族成员falcipain-2、消化泡里的疟原虫富组氨酸蛋白PfHRP Ⅱ、嘌呤核苷磷酸化酶(PfPNP)、肽脱甲酞基化酶(PfPDF)和核糖-5-磷酸异构酶(PfRpiA)等. 近来有研究报道, 在坦桑尼亚实地考察各种抗药性改变的菌株中, Pfcrt 76位和Pfmdr1 86位, Pfdhfr 的51, 59和108位有部分病例存在突变, 但是PfATP6的263和769位都没有发现突变[55]. 另外, 这些在疟原虫中预测的青蒿素作用靶点, 如PfATP6, TCTP, PGH1在酵母菌中都有类似物, 敲除酵母菌中编码这些蛋白的同源基因并检测这些菌株对青蒿素的敏感性, 结果发现除mdl2Δ和pmr1Δ在非发酵培养基中不能正常生长、不能检测青蒿素敏感性以外, 其他的菌株都没有表现出比野生型对照更敏感的表型; 进而在酵母菌中过表达MDL2和PMR1基因, 菌株也没有表现出对青蒿素的抗性, 推测这些靶点在青蒿素抑疟过程中可能不是唯一的或者不是发挥主要作用的[56].
(3) 线粒体模型. 疟原虫的生长需要有功能的线粒体. 加入阿托伐醌(Atovaquone)(一种抗疟药物), 能抑制呼吸链复合物Ⅲ的活性, 破坏疟原虫线粒体功能从而导致虫体死亡, 达到抑疟效果[57]. 在建立了用酵母菌进行青蒿素作用机制研究的模型后, 实验发现, 青蒿素可以直接作用于疟原虫线粒体[58]. 青蒿素的加入可以诱导疟原虫和酵母菌的线粒体肿胀, 产生大量的ROS, 引起线粒体的去极化和膜电势降低, 损害线粒体功能, 但青蒿素对鼠肝细胞的线粒体就没有这样的作用. 当加入ROS 清除剂, 如DPPD 、依达拉奉(edaravone)时, 青蒿素的抗疟和去极化作用能被拮抗, 其他一些ROS 清除剂, 如二硫苏糖醇(dithiothreitol)、α-生育酚(alpha-tocopherol)与青蒿素的拮抗作用也有报道[29]. 另外, 采用两种不同的线粒体通透性转移孔抑制剂均可以拮抗青蒿素的抑疟效果[59]. 根据线粒体模型, 青蒿素可能不是直接作用于某一个蛋白而使线粒体失活, 而可能是通过激活产生ROS 或更多非特异破坏性自由基对细胞造成伤害, 也有可能涉及多个代谢通路, 但目前对其具体的作用过程尚不清楚, 还需更多的实验进行探索.
也有研究对线粒体模型表示质疑[60]. 使用过氧化物处理红内期疟原虫后, 未观察到线粒体形态上的明显变化[61]. 当用荧光染料LysoSensor Blue 和Rhodamine 123分别标记食物泡和线粒体时, 可以在4 h 后观察到青蒿素对食物泡的破坏, 但线粒体没有变化, 从而认为线粒体丧失功能并不是青蒿素类药物发挥作用的早期事件. 但是, 也有较早期的报道称, 对感染了帕氏鼠疟原虫的小鼠施用[3H]-二氢青蒿素,
孙辰等: 青蒿素类药物的作用机制: 一个长久未决的基础研究挑战
350
可以在30 min 时观察到线粒体的膨大[62]. 另有报道, 线粒体形态上的变化是早期事件[63,64]. 出现这些不同的结果可能是实验灵敏度的差别所致, 显微镜下形态学上没有检测到明显的线粒体变化并不能说明其功能没有发生变化.
(4) 其他假说. 正如很多寄生虫一样, 疟原虫体存在着较丰富的还原型谷胱甘肽(GSH), 当虫体食物泡分解血红蛋白时, GSH 能够帮助虫体清除代谢过程中产生的过氧化物和自由基, 实现解毒功能[65]. Wang 和Wu [66]分离得到了青蒿素与GSH 在少量Fe 催化下的复合物, 该发现对研究青蒿素的抗疟机制有提示作用. 此外, 还有一些研究认为青蒿素作用影响了疟原虫膜的合成、辅酶A 的功能、抑制细胞色素酶等, 但是支持这些猜测的实验数据相对较少.
3 青蒿素类药物的抗肿瘤作用
近年来, 有关青蒿素类药物抗肿瘤的研究日益增多. 青蒿素类药物不仅在疟疾治疗方面效果显著, 还可以在体外选择性地抑制多种肿瘤细胞, 如白血病、结肠癌、鼻咽癌和宫颈癌细胞等, 对黑色素瘤、 乳腺癌、前列腺癌等也有一定的抑制作用, 毒副作用较小[67], 且与经典的化疗药物无交叉耐药性. 因而, 有关青蒿素类药物的抗肿瘤机制的研究以及基于此开发抗肿瘤新药成为青蒿素研究的热点.
青蒿素及其衍生物可以在一定程度上抑制肿瘤细胞的增殖. 目前, 提出的抗肿瘤机制主要有: ① 与铁反应产生自由基, 发生烷基化等导致细胞毒性; ② 诱导细胞凋亡[68]; ③ 损伤肿瘤细胞线粒体; ④ 调控肿瘤相关基因的表达量[69]; ⑤ 延迟或抑制细胞周期, 影响肿瘤细胞的DNA 合成[70]; ⑥ 影响免疫系统[71]; ⑦ 抑制肿瘤新生血管的生成[72]等. 当然, 这一过程也有可能是多环节、多通路的, 青蒿素类药物究竟是如何作用于肿瘤细胞仍需更多的研究和探索, 特别是青蒿素抑制肿瘤细胞和抑制疟原虫是否通过同一机制仍不清楚.
在青蒿素抗疟机制的研究中, 有观点认为铁参与了青蒿素的激活和作用过程, 因而也有研究认为, 肿瘤细胞中也存在类似的作用过程, 即铁(尤其是Fe 2+)能与青蒿素及其衍生物作用活化双氧桥, 产生ROS 或发生烷基化作用, 从而破坏细胞功能, 抑制细胞的生长, 同时由于肿瘤细胞中的铁含量高于正常细胞, 因而还可以解释青蒿素类药物对肿瘤细胞的选择性抑制. 实验显示, 肿瘤细胞表面的铁转运蛋白和其受体(transferrin receptor)比正常细胞高出数倍[73]. 有研究认为, 青蒿素类药物与铁相互作用是其抑制肿瘤细胞的上游和关键步骤, 肿瘤细胞的代谢较正常细胞旺盛, 这一过程需要较多的铁, 尤其是二价铁的参与. 实验发现, 青蒿素在有转铁蛋白的条件下对白血病细胞具有更好的选择性[74]; 胞内的heme 可以介导青蒿素的细胞毒性, 增加heme 合成的前体物质会增强青蒿素作用, 而添加抑制剂干扰heme 合成通路则会降低细胞毒性, 若添加非血红素的铁则可通过参与血红素合成过程增强青蒿素的作用[75,76]; 研究显示, 线粒体的呼吸活性对青蒿素类药物的细胞毒性的发挥起关键作用, 当使用HeLa ρ0细胞(没有线粒体DNA 的HeLa 细胞)时, 青蒿琥酯对细胞的抑制作用和细胞毒性会减弱很多[76].
4 总结
上述各种观点和假说表明, 青蒿素类药物的作用机制相当复杂, 可能涉及不同的作用分子, 这些作用有可能协同也可能竞争/拮抗. 随着研究手段的不断发展, 寻找易于操作的、接近于体内环境下疟原虫的生物模型, 如酵母菌模型等, 减少体内体外实验结果不一致的现象, 抑或直接针对疟原虫进行更多分子水平上的操作等, 将有助于推进对青蒿素类药物抗疟作用机制的研究. 对已经提出的模型之间的生理学联系, 还需进一步探索. 尽管, 目前青蒿素仍在抗疟一线发挥着关键的作用, 但人们已经意识到青蒿素抗性病原株出现的风险在日益提高. 破解青蒿素类药物的抗疟机制, 将对新药设计研发和人们的健康有重要意义.
参考文献
1 World Health Organization. World Malaria Report: 2011. XI. Switzerland: WHO Press, 2011
2 Dondorp A M, Nosten F, Yi P, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med, 2009, 361: 455–467
3 青蒿素结构研究协作组. 一种新型的倍半萜内酯——青蒿素. 科学通报, 1977, 3: 142
中国科学: 生命科学 2012年第42卷第5期
4刘静明, 倪慕云, 樊菊芬, 等. 青蒿素(Arteannuin)的结构和反应. 化学学报, 1979, 37: 129–143
5O’Neill P M, Searle N L, Kan K W, et al. Novel, potent, semisynthetic antimalarial carba analogues of the first-generation 1,2,4-trioxane artemether. J Med Chem, 1999, 42: 5487–5493
6Posner G H, Parker M H, Northrop J, et al. Orally active, hydrolytically stable, semisynthetic, antimalarial trioxanes in the artemisinin family. J Med Chem, 1999, 42: 300–304
7Ma J, Weiss E, Kyle D E, et al. Acid catalyzed Michael additions to artemisitene. Bioorg Med Chem Lett, 2000, 10: 1601–1603
8O’Neill P M, Rawe S L, Borstnik K, et al. Enantiomeric 1, 2, 4-trioxanes display equivalent in vitro antimalarial activity versus Plasmodium falciparum malaria parasites: implications for the molecular mechanism of action of the artemisinins. Chem Bio Chem, 2005, 6: 2048–2054 9Liu Y, Cui K, Lu W, et al. Synthesis and antimalarial activity of novel dihydro-artemisinin derivatives. Molecules (Basel, Switzerland), 2011, 16: 4527–4538
10Vennerstrom J L, Arbe-Barnes S, Brun R, et al. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature, 2004, 430: 900–904
11Meshnick S R, Yang Y Z, Lima V, et al. Iron-dependent free radical generation from the antimalarial agent artemisinin (qinghaosu).
Antimicrob Agents Chemother, 1993, 37: 1108–1114
12Eckstein-Ludwig U, Webb R J, Van Goethem I D, et al. Artemisinins target the SERCA of Plasmodium falciparum. Nature, 2003, 424: 957–961
13Wu Y, Yue Z Y, Wu Y L. Interaction of qinghaosu (artemisinin) with cysteine sulfhydryl mediated by traces of non-heme iron. Angew Chem Int Ed Engl, 1999, 38: 2580–2582
14Posner G H, Oh C H, Wang D, et al. Mechanism-based design, synthesis, and in vitro antimalarial testing of new 4-methylated trioxanes structurally related to artemisinin: the importance of a carbon-centered radical for antimalarial activity. J Med Chem, 1994, 37: 1256–1258 15Butler A R, Gilbert B C, Hulme P, et al. EPR evidence for the involvement of free radicals in the iron-catalysed decomposition of qinghaosu (artemisinin) and some derivatives; antimalarial action of some polycyclic endoperoxides. Free Radical Res, 1998, 28: 471–476
16Jefford C W, Vicente M G H, Jacquier Y, et al. The Deoxygenation and isomerization of artemisinin and artemether and their relevance to antimalarial action. Helv Chim Acta, 1996, 79: 1475–1487
17Haynes R K, Chan W C, Lung C M, et al. The Fe2+-mediated decomposition, PfATP6 binding, and antimalarial activities of artemisone and other artemisinins: the unlikelihood of C-centered radicals as bioactive intermediates. Chem Med Chem, 2007, 2: 1480–1497
18O’Neill P M, Bishop L P, Searle N L, et al. Biomimetic Fe(II)-mediated degradation of arteflene (Ro-42-1611). The first EPR spin-trapping evidence for the previously postulated secondary carbon-centered cyclohexyl radical. J Org Chem, 2000, 65: 1578–1582
19Haynes R K, V onwiller S C. The behaviour of qinghaosu (artemisinin) in the presence of non-heme iron(II) and (III). Tetrahedron Lett, 1996, 37: 257–260
20Wu W M, Wu Y k, Wu Y L, et al. Unified mechanistic framework for the Fe(II)-induced cleavage of Qinghaosu and derivatives/analogues.
The first spin-trapping evidence for the previously postulated secondary C-4 radical. J Am Chem Soc, 1998, 120: 3316–3325
21Golenser J, Domb A, Leshem B, et al. Iron chelators as drugs against malaria pose a potential risk. Redox Rep, 2003, 8: 268–271
22Hong Y L, Yang Y Z, Meshnick S R. The interaction of artemisinin with malarial hemozoin.Mol Biochem Parasitol, 1994, 63: 121–128
23Efferth T. Willmar schwabe award 2006: antiplasmodial and antitumor activity of artemisinin—from bench to bedside. Planta Med, 2007, 73: 299–309
24Meshnick S R, Thomas A, Ranz A, et al. Artemisinin (qinghaosu): the role of intracellular hemin in its mechanism of antimalarial action.
Mol Biochem Parasitol, 1991, 49: 181–189
25Li W, Mo W, Shen D, et al. Yeast model uncovers dual roles of mitochondria in action of artemisinin.PLoS Genet, 2005, 1: e36
26王娟, 周兵. 线粒体呼吸链抑制剂对青蒿素代谢速率的影响. 清华大学学报(自然科学版), 2010, 50: 944–946
27Haynes R K, Ho W Y, Chan H W, et al. Highly antimalaria-active artemisinin derivatives: biological activity does not correlate with chemical reactivity. Angew Chem Int Ed Engl, 2004, 43: 1381–1385
28Klonis N, Crespo-Ortiz M P, Bottova I, et al. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion.
Proc Natl Acad Sci USA, 2011, 108: 11405–11410
29Krungkrai S R, Yuthavong Y. The antimalarial action on Plasmodium falciparum of qinghaosu and artesunate in combination with agents which modulate oxidant stress. Trans R Soc Trop Med Hyg, 1987, 81: 710–714
30Levander O A, Ager A L Jr, Morris V C, et al. Qinghaosu, dietary vitamin E, selenium, and cod-liver oil: effect on the susceptibility of mice to the malarial parasite Plasmodium yoelii. Am J Clin Nutr, 1989, 50: 346–352
31Senok A C, Nelson E A, Li K, et al. Thalassaemia trait, red blood cell age and oxidant stress: effect on Plasmodium falciparum growth and
351
孙辰等: 青蒿素类药物的作用机制: 一个长久未决的基础研究挑战
352 sensitivity to artemisinin. Trans R Soc Trop Med Hyg, 1997, 91: 585–589
32Scott M D, Meshnick S R, Williams R A, et al. Qinghaosu-mediated oxidation in normal and abnormal erythrocytes. J Lab Clin Med, 1989, 114: 401–406
33Berman P A, Adams P A. Artemisinin enhances heme-catalysed oxidation of lipid membranes. Free Radic Biol Med, 1997, 22: 1283–1288 34Robert A, Coppel Y, Meunier B. Alkylation of heme by the antimalarial drug artemisinin. Chem Commun(Camb), 2002: 414–415
35Pandey A V, Tekwani B L, Singh R L, et al. Artemisinin, an endoperoxide antimalarial, disrupts the hemoglobin catabolism and heme detoxification systems in malarial parasite. J Biol Chem, 1999, 274: 19383–19388
36Kannan R, Kumar K, Sahal D, et al. Reaction of artemisinin with haemoglobin: implications for antimalarial activity. Biochem J, 2005, 385: 409–418
37Robert A, Benoit-Vical F, Claparols C, et al. The antimalarial drug artemisinin alkylates heme in infected mice. Proc Natl Acad Sci USA, 2005, 102: 13676–13680
38Meunier B, Robert A. Heme as trigger and target for trioxane-containing antimalarial drugs. Acc Chem Res, 2010, 43: 1444–1451
39Kannan R, Sahal D, Chauhan V S. Heme-artemisinin adducts are crucial mediators of the ability of artemisinin to inhibit heme polymerization. Chem Biol, 2002, 9: 321–332
40Loup C, Lelievre J, Benoit-Vical F, et al. Trioxaquines and heme-artemisinin adducts inhibit the in vitro formation of hemozoin better than chloroquine. Antimicrob Agents Chemother, 2007, 51: 3768–3770
41Cazelles J, Robert A, Meunier B. Alkylating capacity and reaction products of antimalarial trioxanes after activation by a heme model. J Org Chem, 2002, 67: 609–619
42Asawamahasakda W, Ittarat I, Chang C C, et al. Effects of antimalarials and protease inhibitors on plasmodial hemozoin production. Mol Biochem Parasitol, 1994, 67: 183–191
43Haynes R K, Monti D, Taramelli D, et al. Artemisinin antimalarials do not inhibit hemozoin formation. Antimicrob Agents Chemother, 2003, 47: 1175
44Coghi P, Basilico N, Taramelli D, et al. Interaction of artemisinins with oxyhemoglobin Hb-FeII, Hb-FeII, carboxyHb-FeII, heme-FeII, and carboxyheme FeII: significance for mode of action and implications for therapy of cerebral malaria. Chem Med Chem, 2009, 4: 2045–2053 45Meshnick S R. Artemisinin and heme. Antimicrob Agents Chemother, 2003, 47: 2712; author reply 2712–2713
46Uhlemann A C, Cameron A, Eckstein-Ludwig U, et al. A single amino acid residue can determine the sensitivity of SERCAs to artemisinins. Nat Struct Mol Biol, 2005, 12: 628–629
47Garah F B, Stigliani J L, Cosledan F, et al. Docking studies of structurally diverse antimalarial drugs targeting PfATP6: no correlation between in silico binding affinity and in vitro antimalarial activity. Chem Med Chem, 2009, 4: 1469–1479
48Jambou R, Legrand E, Niang M, et al. Resistance of Plasmodium falciparum field isolates to in-vitro artemether and point mutations of the SERCA-type PfATPase6. Lancet, 2005, 366: 1960–1963
49Cojean S, Hubert V, Le Bras J, et al. Resistance to dihydroartemisinin. Emerg Infect Dis, 2006, 12: 1798–1799
50Jefford C W. New developments in synthetic peroxidic drugs as artemisinin mimics. Drug Discov Today, 2007, 12: 487–495
51Valderramos S G, Scanfeld D, Uhlemann A C, et al. Investigations into the role of the Plasmodium falciparum SERCA (PfATP6) L263E mutation in artemisinin action and resistance. Antimicrob Agents Chemother, 2010, 54: 3842–3852
52Cui L, Wang Z, Jiang H, et al. Lack of association of the S769N mutation in plasmodium falciparum SERCA (PfATP6) with resistance to artemisinins. Antimicrob Agents Chemother, 2012, 56: 2546–2552
53Cardi D, Pozza A, Arnou B, et al. Purified E255L mutant SERCA1a and purified PfATP6 are sensitive to SERCA-type inhibitors but insensitive to artemisinins. J Biol Chem, 2010, 285: 26406–26416
54Arnou B, Montigny C, Morth J P, et al. The Plasmodium falciparum Ca(2+)-ATPase PfATP6: insensitive to artemisinin, but a potential drug target. Biochem Soc Trans, 2011, 39: 823–831
55Kamugisha E, Jing S, Minde M, et al. Efficacy of artemether-lumefantrine in treatment of malaria among under-fives and prevalence of drug resistance markers in Igombe-Mwanza, north-western Tanzania. Malar J, 2012, 11: 58
56黄倩, 周兵. 酵母菌中青蒿素作用靶点的探索. 清华大学学报(自然科学版), 2008, 48: 408–411
57Srivastava I K, Rottenberg H, Vaidya A B. Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in
a malarial parasite. J Biol Chem, 1997, 272: 3961–3966
58Wang J, Huang L, Li J, et al. Artemisinin directly targets malarial mitochondria through its specific mitochondrial activation. PLoS One, 2010, 5: e9582
59王娟, 黄丽英, 龙伊成, 等. 线粒体通透性转移孔与青蒿素抗疟机制研究. 现代生物医学进展, 2009, 9: 4006–4009
中国科学: 生命科学 2012年第42卷第5期
60del Pilar Crespo M, Avery T D, Hanssen E, et al. Artemisinin and a series of novel endoperoxide antimalarials exert early effects on digestive vacuole morphology. Antimicrob Agents Chemother, 2008, 52: 98–109
61Afonso A, Hunt P, Cheesman S, et al. Malaria parasites can develop stable resistance to artemisinin but lack mutations in candidate genes atp6 (encoding the sarcoplasmic and endoplasmic reticulum Ca2+ ATPase), tctp, mdr1, and cg10. Antimicrob Agents Chemother, 2006, 50: 480–489
62Ellis D S, Li Z L, Gu H M, et al. The chemotherapy of rodent malaria, XXXIX. Ultrastructural changes following treatment with artemisinine of Plasmodium berghei infection in mice, with observations of the localization of [3H]-dihydroartemisinine in P. falciparum in vitro. Ann Trop Med Parasit, 1985, 79: 367–374
63Maeno Y, Toyoshima T, Fujioka H, et al. Morphologic effects of artemisinin in Plasmodium falciparum. Am J Trop Med Hyg, 1993, 49: 485–491
64Kawai S, Kano S, Suzuki M. Morphologic effects of artemether on Plasmodium falciparum in Aotus trivirgatus. Am J Trop Med Hyg, 1993, 49: 812–818
65Linares G E, Rodriguez J B. Current status and progresses made in malaria chemotherapy. Curr Med Chem, 14: 289–314
66Wang D Y, Wu Y L. A possible antimalarial action mode of qinghaosu (artemisinin) series compounds. Alkylation of reduced glutathione by C-centered primary radicals produced from antimalarial compound qinghaosu and 12-(2,4-dimethoxyphenyl)-12-deoxoqinghaosu. Chem Commun, 2000, 2193–2194
67Efferth T, Dunstan H, Sauerbrey A, et al. The anti-malarial artesunate is also active against cancer. Int J Oncol, 2001, 18: 767–773
68Gao N, Budhraja A, Cheng S, et al. Interruption of the MEK/ERK signaling cascade promotes dihydroartemisinin-induced apoptosis in vitro and in vivo. Apoptosis, 2011, 16: 511–523
69Efferth T, Olbrich A, Bauer R. mRNA expression profiles for the response of human tumor cell lines to the antimalarial drugs artesunate, arteether, and artemether. Biochem Pharmacol, 2002, 64: 617–623
70Zhao Y, Jiang W, Li B, et al. Artesunate enhances radiosensitivity of human non-small cell lung cancer A549 cells via increasing NO production to induce cell cycle arrest at G2/M phase. Int Immunopharmacol, 2011, 11: 2039–2046
71Noori S, Hassan Z M. Dihydroartemisinin shift the immune response towards Th1, inhibit the tumor growth in vitro and in vivo. Cell Immunol, 2011, 271: 67–72
72Zhou H J, Wang W Q, Wu G D, et al. Artesunate inhibits angiogenesis and downregulates vascular endothelial growth factor expression in chronic myeloid leukemia K562 cells. Vascul Pharmacol, 2007, 47: 131–138
73Efferth T, Benakis A, Romero M R, et al. Enhancement of cytotoxicity of artemisinins toward cancer cells by ferrous iron. Free Radic Biol Med, 2004, 37: 998–1009
74Lai H, Sasaki T, Singh N P, et al. Effects of artemisinin-tagged holotransferrin on cancer cells. Life Sci, 2005, 76: 1267–1279
75Zhang S, Gerhard G S. Heme mediates cytotoxicity from artemisinin and serves as a general anti-proliferation target. PLoS One, 2009, 4: e7472
76Mercer A E, Copple I M, Maggs J L, et al. The role of heme and the mitochondrion in the chemical and molecular mechanisms of mammalian cell death induced by the artemisinin antimalarials. J Biol Chem, 2011, 286: 987–996
353
孙辰等: 青蒿素类药物的作用机制: 一个长久未决的基础研究挑战
354 Mechanism of Action of Artemisinins: a Long Unsettled Challenge
SUN Chen, LI Jian & ZHOU Bing
State Key Laboratory of Biomembrane and Membrane Biotechnology, School of Life Sciences, Tsinghua University, Beijing 100084, China
Artemisinin, discovered by Chinese scientists in the early 1970s, is an effective antimalarial drug with low toxicity. Artemisinins (artemisinin and its derivatives) remain as the gold standard in combating ever increasing drug-resistant malaria. Although intensive efforts have been devoted to explore the mode of action of this class of drugs, its exact mechanism remains an enigma. Many hypotheses have been proposed, such as iron and heme model, mitochondria model, PfATP6 model, and so forth. Recent studies have demonstrated that artemisinin and its analogs also possess potent inhibitory activities on some other parasites, cancer, virus and rheumatism. The aim of this review is to provide a mechanistic overview of the action of artemisinins.
artemisinin, mechanism, heme, mitochondria
doi: 10.1360/052012-168
周兵, 教授, 博士生导师. 1987年毕业于复旦大学, 1988年通过CUSBEA项
目留学美国伊利诺伊大学香槟分校(University of Illinois, Urbana-Champaign),
1989~1995年就读于加州大学伯克利分校, 获得分子细胞学博士学位, 2002年于
加州大学旧金山医学院完成博士后学习, 同年入选“百人计划”被引进清华大学
生命科学学院任职. 2006年获得国家杰出青年科学基金资助. 现从事青蒿素作
用机理以及微量金属元素代谢/功能的研究.
本文为Science China Life Sciences CUSBEA article series专栏特邀文章.
——编者注
青蒿素性质及合成方法 院系:化工学院 专业:应用化学 学号: 姓名: 指导老师: 2016/1/12 摘要:青蒿素是目前治疗疟疾的特效药。本文对自青蒿素发现以来的最新研究进展进行了比较详尽的综述。内容包括:青蒿素的特性,青蒿素的合成,青蒿素的生物合成,青蒿素衍生物。 关键词:青蒿素;合成方法;青蒿素衍生物 Abstract:The recent research advances in artemisinin, the most effective weapons against malarial parasites have been reviewed. An overview is given on artemisinin research from the following aspects:sources of artemisinin,synthesisof artemisinin, biosynthesis of artemisinin, analogs of artemisinin and artemisinin production from plant tissue cultures。 Key words:artemisinin,synthesis,artemisinin derivatives 目录 1、前言……………………………………………………………… 2、青蒿素的基本性质………………………………………………
(1)分子结构………………………………………………………… (2)理化性质………………………………………………………… (3)药动力…………………………………………………………… (4)提取工艺………………………………………………………… 3、合成方法………………………………………………………… (1)全合成………………………………………………………… (2)半合成………………………………………………………… (3)生物合成……………………………………………………… 4、衍生物………………………………………………………… 5、抗癌功能………………………………………………………… 6.结论……………………………………………………………… 1前言: 青蒿素是中国学者在20世纪70年代初从中药黄花蒿( Artem isia annua L1 )中分离得到的抗疟有效单体化合物,是目前世界上最有效的治疗脑型疟疾和抗氯喹恶性疟疾的药物, 对恶性疟、间日疟都有效, 可用于凶险型疟疾的抢救和抗氯喹病例的治疗。青蒿素还具有抑制淋巴细胞的增殖和细胞毒性的用;具有影响人体白血病U937细胞的凋亡及分化的作用;还具有部分逆转MCF-7/ARD细胞耐药性作用;还具有抑制人胃癌裸鼠移植瘤的生长的作用;还具有一定的抗肿瘤作用等。除此之外,青蒿素及其衍生物还具有生物抗炎免疫作用、生物抗肿瘤作用、抑制神经母细胞瘤细胞增殖的作用等。世界卫生组织确定为治疗疟疾的首选药物, 具有快速、高效、和低毒副作用的特征。因在发现青蒿素过程中的杰出贡献,屠呦呦先后被授予2011年度拉斯克临床
青蒿素的发现,提取及一系列发展应用 1.时代背景:时代背景.mp4 世界上影响人数最多的疾病并非现在深受关注的艾滋病,而是一种堪称“历史悠久”的疾病——疟疾,也就是俗称的“打摆子”,同时,它也是当今除艾滋病外,上升趋势最为显著的一种传染病,每年2~3亿人感染此病,200多万人死亡。19世纪从南美洲金鸡纳树皮中得到的奎宁曾成为最有效的药物,治愈了众多的疟疾患者。20世纪第二次世界大战后模仿奎宁基本结构而合成的一批新药如氯喹、伯喹也曾救治过无数的病人。但是20世纪60年代出现抗药性疟原虫后,以往常用的抗疟药(如氯喹、磺胺、奎宁等)的效果便不复存在,以至于造成了无药可医的局面,特别在东南亚、非洲地区情况更为严重。青蒿素类药物的出现以其副作用低且不易产生抗药性而被誉为“治疗疟疾的最大希望”。 2. 什么是青蒿素时代背景.mp4 ◆分子式为C15H22O5,分子量282.33,组分含量:C 63.81%,H 7.85%,O 28.33%。 ◆无色针状晶体,味苦。 ◆在丙酮、醋酸乙酯、氯仿、苯及冰醋酸中易溶,在乙醇和甲醇、乙醚及石油醚中可溶解,在水中几乎不溶。
青蒿素(Artemisinin)又名黄蒿素,是一种具有过氧桥的倍半萜内酯类化合物。分子式为C15H22O5,分子量为282.34,具有过氧键和δ-内酯环,有一个包括氧化物在内的1,2,4-三恶烷结构单元,在自然界中是非常罕见的,它的分子中包括7个手性中心。青蒿素为无色针状结晶,熔点为156~157℃,易溶于氯仿、丙酮、乙酸乙酯和苯,
可溶于乙醇、乙醚,微溶于冷石油醚,几乎不溶于水。因其具有特殊的过氧基团,对热不稳易受湿、热和还原性物质的影响而分解。 3.为什么要选用青蒿治疗疟疾? 疟疾是一个非常古老的疾病。我们的先人对它还是有一定办法的。在晋代葛洪所著的《肘后备急方》中就有关于疟疾的治疗方药,原文如下:青蒿一握,以水二升渍,绞取汁,尽服之。意思是,用一把青蒿,以二升的水浸渍以后,绞扭青蒿,取得药汁,然后一次服尽。可别小看这几句话,它说明,我们的古人对于青蒿截疟已经有了很深入的认识。 4.验证青蒿素对疟疾的治疗效果实验: 为什么在实验室里青蒿的提取物不能很有效地抑制疟疾呢?是提取方法有问题?还是做实验的老鼠有问题? “青蒿一握,以水二升渍,绞取汁,尽服之”为什么这和中药常用的高温煎熬法不同?原来古人用的是青蒿鲜汁!温度!这两者的差别是温度!很有可能在高温的情况下,青蒿的有效成分就被破坏掉了。改用沸点较低的乙醚进行实验,她在60摄氏度下制取青蒿提取物。接下来在实验室里,青蒿提取物对疟原虫的抑制率达到了100%!
服用他汀类药物6注意 他汀类药物是当前最常用的调脂药物。我国已逐渐步入老年化社会,心脑血管疾病发病率呈不断上升的趋势,血脂异常与心脑血管疾病的发生、发展和预后均密切相关。使用他汀类药物(包括辛伐他汀、普伐他汀、洛伐他丁、阿托伐他汀、瑞舒伐他汀等),既能够调节血脂水平,又能稳定人体血管里的潜在病灶——粥样斑块(稳斑块),从而能有效降低心脑血管疾病的发生率。不过,使用他汀类药物需要注意以下几点—— 1.用药疗程:对于已有心脑血管疾病發生(如脑中风)的患者,或者有高危因素(如糖尿病高血压)患者,血脂水平即使在正常范围,也需要在医生指导下服用此类药以预防疾病再次发作,长期坚持服药才能有效。 2.服用剂量:他汀类药物所致肝损害的发生与服用剂量有一定的
关系。服用10~20毫克的剂量,一般不引起转氨酶升高;但如剂量 增加至40毫克或大剂量80毫克时,就会出现以转氨酶升高为特征的肝损害。因此请记住,不能擅自增加服用剂量。 3.肌肉疼痛:极少数患者服药后会出现无法解释的肌肉疼痛不适、肌肉酸软、僵直或痉挛,如有此类情况发生,请来医院就诊。 4.肝损害:极少数患者服药后会出现转氨酶升高,如果检查肝功能发现有此异常,如果转氨酶超过正常值上限的3倍,需要停药就诊。建议初次服药后1~2月复查肝功能,如果没有异常,可半年或1年 复查肝功能1次。还有极少数患者会有肝炎临床症状,如出现纳差、乏力、腹胀、恶心、呕吐、黄疸、肝痛,此时应立即停药就诊。 5.联合用药:联合用药是他汀类药物所致肝损害的重要危险因素。如果同时服用氯吡格雷、阿奇霉素、胺碘酮、罗红霉素、非诺贝特、氟他胺、曲格列酮等药,由于肝脏代谢减慢,可促进他汀类药物的血药浓度,从而增加副作用。因此,服用他汀类药物患者在就诊时一定要告诉医生自己正在服用哪些药,并注意有无上述不良反应的发生。
抗肿瘤药物的作用机制 1.细胞生物学机制 几乎所有的肿瘤细胞都具有一个共同的特点,即与细胞增殖有关的基因被开启或激活,而与细胞分化有关的基因被关闭或抑制,从而使肿瘤细胞表现为不受机体约束的无限增殖状态。从细胞生物学角度,诱导肿瘤细胞分化,抑制肿瘤细胞增殖或者导致肿瘤细胞死亡的药物均可发挥抗肿瘤作用。 2.生化作用机制 (1)影响核酸生物合成:①阻止叶酸辅酶形成;②阻止嘌呤类核苷酸形成;③阻止嘧啶类核苷酸形成;④阻止核苷酸聚合;(2)破坏DNA结构和功能;(3)抑制转录过程阻止RNA 合成;(4)影响蛋白质合成与功能:影响纺锤丝形成;干扰核蛋白体功能;干扰氨基酸供应;(5)影响体内激素平衡。 烷化剂烷化剂可以进一步分为: 氮芥类:均有活跃的双氯乙基集团,比较重要的有氮芥、苯丁酸氮芥、环磷酰胺(CTX)、异环磷酰胺(IFO)等。其中环磷酰胺为潜伏化药物需要活化才能起作用。目前临床广泛用于治疗淋巴瘤、白血病、多发性骨髓瘤,对乳腺癌、肺癌等也有一定的疗效。 该药除具有骨髓抑制、脱发、消化道反应,还可以引起充血性膀胱炎,病人出现血尿,临床在使用此药时应鼓励病人多饮水,达到水化利尿,减少充血性膀胱炎的发生。还可以配合应用尿路保护剂美斯纳。 亚硝脲类:最早的结构是N-甲基亚硝脲(MNU)。以后,合成了加入氯乙集团的系列化合物,其中临床有效的有ACNU、BCNU、CCNU、甲基CCNU等,链氮霉素均曾进入临床,但目前已不用。其中ACNU、BCNU、CCNU、能通过血脑屏障,临床用于脑瘤及颅内转移瘤的治疗。主要不良反应是消化道反应及迟发性的骨髓抑制,应注意对血象`的观测,及时发现给予处理。 乙烯亚胺类:在研究氮芥作用的过程中,发现氮芥是以乙烯亚胺形式发挥烷化作用的,因此,合成了2,4,6-三乙烯亚胺三嗪化合物(TEM),并证明在临床具有抗肿瘤效应,但目前在临床应用的只有塞替派。此药用于治疗卵巢癌、乳腺癌、膀胱癌,不良反应主要为骨髓抑制,注意对血象定期监测。 甲烷磺酸酯类:为根据交叉键联系之复合成的系列化合物,目前临床常用的只有白消安(马利兰)。临床上主要用于慢性粒细胞白血病,主要不良反应是消化道反应及骨髓抑制,个别病人可引起纤维化为严重的不良反应。遇到这种情况应立即停药,更换其它药物。 其他:具有烷化作用的有达卡巴嗪(DTIC)、甲基苄肼(PCZ)六甲嘧胺(HHN)等。环氧化合物,由于严重不良反应目前已被淘汰。 抗代谢药物抗代谢类药物作用于核酸合成过程中不同的环节,按其作用可分为以下几类药物: 胸苷酸合成酶抑制剂:氟尿嘧啶(5-FU)、呋喃氟尿嘧啶(FT-207)、二喃氟啶(双呋啶FD-1)、优氟泰(UFT)、氟铁龙(5-DFUR)。 抗肿瘤作用主要由于其代谢活化物氟尿嘧啶脱氧核苷酸干扰了脱氧尿嘧啶苷酸向脱氧胸腺嘧啶核苷酸转变,因而影响了DNA的合成,经过四十年的临床应用,成为临床上常用的抗肿瘤药物,成为治疗肺癌、乳腺癌、消化道癌症的基本药物。 不良反应比较迟缓,用药6-7天出现消化道粘膜损伤,例如:口腔溃疡、食欲不振、恶心、呕吐、腹泻等,一周以后引起骨髓抑制。而连续96小时以上粘腺炎则成为其主要毒性反应。临床上如长时间连续点滴此类药物应做好病人的口腔护理,教会病人自己学会口腔清洁的方法,预防严重的粘膜炎发生。
不同他汀类药物的分类比较 动脉粥样硬化性心血管病(ASCVD)是导致老年人死亡和影响生活治疗的主要疾病,随着年龄的增长,ASCVD的发病率和死亡率逐渐增加。血脂异常作为ASCVD事件的独立危险因素,控制血脂水平已成为当前预防与治疗ASCVD 的主要方式。临床常规检验提供的血脂参数主要包括总胆固醇(TC)、高密度脂蛋白胆固醇(HDL-C)、低密度脂蛋白胆固醇(LDL-C)、极低密度脂蛋白胆固醇(VLDL-C)与甘油三酯(TG)。其中LDL-C水平与ASCVD的发病风险相关性最大,LDL可通过血管内皮进人血管壁内,在内皮下滞留的LDL被修饰成氧化型LDL,后者被巨噬细胞吞噬后形成泡沫细胞,泡沫细胞不断增多融合,构成动脉粥样硬化斑块的脂质核心。因此,应将LDL-C作为控制血脂异常的主要干预靶点。他汀类药物能明显的降低LDL,显著改善患者预后,具有最充分的临床研究证据,是被国内外各大指南推荐首选的调脂药物。 他汀类药物是羟甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂,可抑制内源性胆固醇合成的限速酶HMG-CoA还原酶降低胆固醇和脂蛋白水平,同时还可增加肝脏细胞表面LDL受体以增强LDL的摄取和分解代谢,减少体内LDL水平,从而通过上述药理机制发挥调脂作用。本院常用的他汀类药物有氟伐他汀(来适可)、普伐他汀(富利他之)、辛伐他汀(舒降之)、阿托伐他汀(阿乐、尤佳、立普妥)和瑞舒伐他汀(可定)等。本文通过不同他汀类药物的作用强度、药动学特征、不良反应等方面来介绍各药物的特点,以保障他汀类药物的合理应用。 1.作用强度:胆固醇治疗的目标值根据患者具体病情需使LDL-C降低至<1.8、 2.6或 3.4mmol/L(详见相关指南)。不同他汀类药物降LDL-C强度差异明显,以LDL-C降幅38%为例,每日需氟伐他汀80mg、普伐他汀40mg、辛伐他汀20mg、阿托伐他汀10mg、瑞舒伐他汀<5mg。结合本院各他汀药物剂量和常规用法用量(富利他之20mg,qn;来适可40mg,qn;舒降之20mg,qn;阿乐、立普妥20mg,qd,尤佳10mg,qd;可定10mg,qd),可推算普伐他汀(富利他之)、氟伐他汀(来适可)具有低强度的降脂作用(使LDL-C降低<30%),而辛伐他汀(舒降之)、阿托伐他汀(阿乐、尤佳、立普妥)和瑞舒伐他汀(可定)具有中等强度的降脂作用(使LDL-C降低30-50%)。可见同等剂量他汀类药物
青蒿素的合成与研究进展 摘要:青蒿素是目前世界上最有效的治疗疟疾的药物之一,存在活性好、毒副作用小、市场需求大、来源窄等特点。目前,青蒿素的获取途径主要有直接从青蒿中提取、化学合成和生物合成。本综述将针对近年来青蒿素的发展特点及合成方法进行论述。 关键词:青蒿素;合成方法;研究进展 青蒿素是中国学者在20世纪70年代初从中药黄花蒿( Artem isia annua L1 )中分离得到的抗疟有效单体化合物,是目前世界上最有效的治疗脑型疟疾和抗氯喹恶性疟疾的药物, 对恶性疟、间日疟都有效, 可用于凶险型疟疾的抢救和抗氯喹病例的治疗。青蒿素还具有抑制淋巴细胞的增殖和细胞毒性的用1;具有影响人体白血病U937细胞的凋亡及分化的作用2;还具有部分逆转MCF-7/ARD细胞耐药性作用3;还具有抑制人胃癌裸鼠移植瘤的生长的作用4;还具有一定的抗肿瘤作用5等。除此之外,青蒿素及其衍生物还具有生物抗炎免疫作用、生物抗肿瘤作用、抑制神经母细胞瘤细胞增殖的作用等。世界卫生组织确定为治疗疟疾的首选药物, 具有快速、高效、和低毒副作用的特征。6。因在发现青蒿素过程中的杰出贡献,屠呦呦先后被授予2011年度拉斯克临床
医学研究奖和2015年诺贝尔医学奖。 1 青蒿素的理化性质及来源 青蒿素的分子式为C15H22O5, 相对分子质量为282. 33。是一种含有过氧桥结构的新型倍半萜内酯,有一个包括过氧化物在内的1,2,4-三烷结构单元,它的分子中还包括7个手性中心,合成难度很大。中国科学院有机所经过研究,解决了架设过氧桥难题,在1983年完成了青蒿素的全合成。青蒿素也有一些缺点, 如在水和油中的溶解度比较小, 不能制成针剂使用等。 2 青蒿中提取青蒿素 青蒿素是从菊科植物黄花蒿中提取出来的含有过氧桥的倍半萜内酯类化合物,在治疗疟疾方面具有起效快、疗效好、使用安全等特点。目前主要的提取方法有溶剂提取法、超临界提取法、超声波萃取法、微波萃取法、其他萃取法等。2.1有机溶剂萃取青蒿素 水蒸气蒸馏(steam distillation,SD)法由于其具有设备简单,操作安全,不污染环境,成本低,避免了提取过程中有机溶剂残留对油质造成影响等特点,是有效提取中药挥发油的重要方法。有机溶剂提取法是目前青蒿中许多有效成分的提取目前仍然常用的方法,常用的溶剂有醇类(甲醇、乙醇
他汀类药物有哪些主要的副作用他汀类药物(statins)是羟甲基戊二酰辅酶A(HMG-CoA)还原酶抑制剂,此类药物通过竞争性抑制内源性胆固醇合成限速酶(HMG-CoA)还原酶,阻断细胞内羟甲戊酸代谢途径,使细胞内胆固醇合成减少,从而反馈性刺激细胞膜表面(主要为肝细胞)低密度脂蛋白(low density lipoprotein,LDL)受体数量和活性增加、使血清胆固醇清除增加、水平降低。他汀类药物还可抑制肝脏合成载脂蛋白B-100,从而减少富含甘油三酯AV、脂蛋白的合成和分泌。 他汀类药物分为天然化合物(如洛伐他丁、辛伐他汀、普伐他汀、美伐他汀)和完全人工合成化合物(如氟伐他汀、阿托伐他汀、西立伐他汀、罗伐他汀、pitavastatin)是最为经典和有效的降脂药物,广泛应用于高脂血症的治疗。 他汀类药物除具有调节血脂作用外,在急性冠状动脉综合征患者中早期应用能够抑制血管内皮的炎症反应,稳定粥样斑块,改善血管内皮功能。延缓动脉粥样硬化(AS)程度、抗炎、保护神经和抗血栓等作用。 结构比较: 辛伐他汀(Simvastatin)是洛伐他汀(Lovastatin)的甲基化衍化物。 美伐他汀(Mevastatin,又称康百汀,Compactin)药效弱而不良反应多,未用于临床。目前主要用于制备它的羟
基化衍化物普伐他汀(Pravastatin)。 体内过程: 洛伐他汀和辛伐他汀口服后要在肝脏内将结构中的其内酯环打开才能转化成活性物质。 相对于洛伐他汀和辛伐他汀,普伐他汀本身为开环羟酸结构,在人体内无需转化即可直接发挥药理作用,且该结构具有亲水性,不易弥散至其他组织细胞,极少影响其他外周细胞内的胆固醇合成。 除氟伐他汀外,本类药物吸收不完全。 除普伐他汀外,大多与血浆蛋白结合率较高。 有效控制血脂成了众多血脂异常患者及家属迫切需要解决的问题。有的患者从某些媒体上、广告上听说了哪种药物好,就自行去购买服用;有些人一听说他汀类药好,就非要医生给他开他汀类药物不可。殊不知,药不对症,即使用了他汀类药物,效果也不一定好,这是什么原因呢?这是因为他汀类药物有它的适应人群,并非人人皆可。 人体血液中脂质一种或几种成分的升高或降低,叫做血脂异常。过去,将其中某些脂质成分的升高称为高脂血症或高脂蛋白血症,现在则统称为血脂异常。
青蒿素综述 刘兵情 (井冈山大学11级药本(1)班学号:111116023) 摘要:青蒿素类抗疟药物的发现是全球抗疟药物发展史上继奎宁之后的又一里程碑[1], 是目前治疗疟疾的特效药.本文简要介绍青蒿素的发现过程、药源、生物合成、应用前景和青蒿素及其衍生物药理活性,重点在于介绍青蒿素生物合成过程。 关键词:青蒿素发现过程药源生物合成药理活性前景 引言:青蒿素是在科研计划组织下,全国多部门、多学科专家尽心协作、相互 配合取得的重大成果,是继承发扬我国传统医药宝库的成功范例[2]。青蒿素主要有抗疟、抗孕、抗纤维化、抗吸血虫等药理作用[3]。青蒿素生物合成三个阶段分为从乙酰辅酶A 到法呢基焦磷酸的“上游”途径、从法呢基焦磷酸到双氢青蒿酸的“中游”途径和从双氢青蒿酸到青蒿素的“下游”途径,其中上游途径青蒿及其他高等植物与酵母等真核微生物完全相同,因而只需在酵母中额外增加一个青蒿素合成代谢支路, 就能让酵母全合成青蒿素。而中游的酶促反应在酵母中已经完全建立,下游途径的反应条件在酵母中则未建立[4]。而且青蒿素及其衍生物在抗肿瘤和葡萄膜炎免疫治疗上也具有应用前景 。 一.青蒿素药物来源 1967 年北京《5·23 抗疟计划》付诸实施, 1969 年1 月北京中医研究院加入 5·23 计划,任命屠呦呦为科研组组长, 在全国多个研究单位协作下, 组织植物化学与药理学等专业200 多人参加, 并与中医药工作者密切合作[5].从追索我国历代抗疟方剂入手, 科研组调查了 2 000 种中草药制剂, 从中选出可能具抗疟活性的达640 种. 余亚纲梳理开列了有808 个中药的单子,其中有乌头、乌梅、鳖甲、青蒿等[6]共用约200种国产草药制成380 多种抽提物, 再筛查它们对小鼠疟疾模型的疗效,但实验不易获得明显结果[7]军事医学科学院用鼠疟模型筛选了近百个药方,青蒿提取物的抑制率虽达60%~80%, 而效力不够稳定[6]继后, 研究组经余亚纲和顾国明复筛, 肯定了青蒿的抗疟作用[8]他们也研究了中药常山,其抗疟作用虽强, 但呕吐的副作用亦强而妨碍推广应用. 转折点出现在黄花蒿的抽提物. 传统中药青蒿包括两个品种: 学名黄花蒿(Artemisia an-nua L.)的抽提物能对小鼠疟原虫的生长显示良好的抑制作用;而学名青蒿(Artemisia apiaceaHance)则无任何抗疟作用[7][9],继后的实验中, 上述结果未能重复, 这同中医文献的记载相矛盾. 为解开此疑惑, 再深入查阅古代医学文献, 最后在晋朝葛洪著《肘后备急方》中找到“青蒿一握, 以水二升渍, 绞取汁, 尽服之”的抗疟记录. 惯常煎熬中药的高温抽提法已破坏了抗疟的活性组分;温度高于60 ℃将使青蒿素完全分解. 在较低温度下进行青蒿抽提后, 获得了很满意的效果[7][9][10]
他汀类药物作用机制 令狐文艳 他汀类药物有明显的调血脂作用,人体内Ch主要来自肝脏合成,在Ch合成过程中HMG-CoA还原酶使HMG-CoA转换为中间产物MVA。他汀类具有与HMG-CoA相似的结构,且和HMG-CoA 还原酶的亲和力高出HMG-CoA数千倍,对该酶发生竞争性抑制,使Ch合成受阻,除使血浆Ch浓度降低外,还通过负反馈调节导致肝细胞表面LDL受体代偿性增加及活性增强,致使血浆LDL降低,继而导致VLDL代谢加快,再加上肝脏合成及释放VLDL减少,也导致VLDL及TG相应下降。HDL的升高,可能是由于VLDL减少的间接结果。由于各种他汀类药物与HMG-CoA还原酶亲和力的不同,所以调脂的效应各异。 比如常用的阿托伐他汀:用于治疗高胆固醇血症和混合型高脂血症;冠心病和脑中风的防治。 本品为他汀类血脂调节药,属HMG-CoA还原酶抑制剂。本身无活性,口服吸收后的水解产物在体内竞争性地抑制胆固醇合成过程中的限速酶羟甲戊二酰辅酶A还原酶,使胆固醇的合成减少,也使低密度脂蛋白受体合成增加,主要作用部位在肝脏,结果使血胆固醇和低密度脂蛋白胆固醇水平降低,中度降低血清甘油三酯水平和增高血高密度脂蛋白水平。由此对动脉粥样硬化和冠心病的防治产生作用。
本品口服吸收良好,因经肝内广泛首关代谢,绝对生物利用度较低,大约为12%,本品在肝脏经细胞色素P4503A4代谢为多种活性代谢物。阿托伐他汀的平均血浆半衰期大约为14小时,但由于其活性代谢物的影响,实际对HMG-CoA还原酶抑制作用的半衰期为20~30小时。本品蛋白结合率为98%,大部分以代谢物的形式经胆汁排出。
青蒿素发明人获诺奖的背后争议 资料来源:新浪新闻中心和腾迅网 中新网10月5日电据诺贝尔奖官网的最新消息,瑞典斯德哥尔摩当地时间5日中午11时30分,2015年诺贝尔生理学或医学奖在当地的卡罗琳斯卡医学院揭晓,爱尔兰医学研究者威廉·坎贝尔、日本学者Satoshi Omura以及中国药学家屠呦呦荣获了该奖项。 2015年10月7日 - 屠呦呦通过央视正式发表获奖感言:获诺奖是一个很大的荣誉,青蒿素研究成功是多年研... 这也是青蒿素成就归属有争论的原因之一。为了平等而取消... 屠呦呦女,生于1930年12月,中国中医研究院终身研究员兼首席研究员,青蒿素研究开发中心主任。1980年聘为硕士生导师,2001年聘为博士生导师。突出贡献是创制新型抗疟药———青蒿素和双氢青蒿素。 青蒿素,用去了屠呦呦大半生时间,她却依然痴迷于此,未曾停歇。她说,“荣誉多了,责任更大,我还有很多事要做。” 屠呦呦:实验191次才成功以身试毒弄坏肝脏 一面是热情和盛赞,另一面就是争议和不解。 1、据说中国领导人是应越南请求才举全国之力攻坚疟疾的,该项目和人工合成牛胰岛素结晶一样,从一开始就是一个政治工程,是一个不小的军事计划的一部分,即全国性抗疟研究计划“523战备项目”,志在帮助北越“打击美帝”。 2、从草药中寻找抗疟成分并不是新鲜主意,1941年,上海第一医学院药理学教授张昌绍就曾尝试利用中药治疗疟疾,1946年
和1948年分别在《科学》和《自然》报道中药常山及其活性成分的抗疟作用。然而张昌绍在文革中被逼自杀,另一些原本致力于此的科学家也被关牛棚、靠边站。所以项目里都是中青年,屠呦呦作为中医研究院中药研究所实习研究员加入进去的时候是38岁。 3、屠呦呦起到的关键作用就是发现粗提取的青蒿素受热就失去活性,想到了要用乙醚提取,据说是受其他研究小组用甲醚提取成功后得到的启发。不过用水、甲醇、汽油、乙醚等各种溶剂去萃取本就是制药的规范步骤,因为当时懂得科研规范的人都靠边站了,所以只能靠屠呦呦拍脑门灵光乍现。 4、青蒿(又名香蒿)中不含青蒿素,含青蒿素的是黄花蒿(又名臭蒿)。作为中药市售的青蒿或多或少混杂黄花蒿,数量不等,导致当时各组临床试验结果不稳定。云南和山东药物研究所提取的青蒿素效果很好,反而屠呦呦组提取的青蒿素不仅无效,还有较明显心脏毒副作用。 5、课题小组在重庆买到假冒伪劣青蒿,全是黄花蒿,于是抑菌率100%。 6、青蒿素主要产自黄花蒿和大头黄花蒿,云南和山东省药物研究所一开始命名为“黄蒿素”和“黄花蒿素”。1978年,523项目的科研成果鉴定会“按中药用药习惯”,“将中药青蒿原植物只保留黄花蒿一种,而其抗疟成分随传统中药定名为青蒿素”。青蒿素的学名还是援引了黄花蒿的拉丁名。 7、卫生部动用了数十个单位的500多名科研人员,用5年
双氢青蒿素抗疟机制 据2018年WHO发布的《世界疟疾报告》估计,2017年全世界共发生2.19亿例疟疾,全球有43.5万人死于疟疾,而2016年全球共有2.16亿疟疾病例,约44.5万人死于疟疾,消除疟疾仍然是一个严峻挑战。目前发现有五种疟原虫会使人类感染疟疾,包括恶性疟原虫(Plasmodium falciparum)、三日疟原虫(Plasmodium malariae)、卵形疟原虫(Plasmodiumovale)、间日疟原虫(Plasmodium vivax)及诺氏疟原虫(Plasmodium knowlesi)。 其中恶性疟原虫引发的疟疾危害最大,死亡率最高。据2018年《世界疟疾报告》,恶性疟原虫是撒哈拉以南非洲最流行的疟疾寄生虫,占2017年估计疟疾病例总数的92%。 以青蒿素为基础的联合疗法是疟疾治疗的主要方法。青蒿素及其衍生物在疟疾治疗上挽救了全球数百万人的生命,但是其抗疟机制仍有待于进一步阐明。 对青蒿素抗疟机制的研究不仅有助于青蒿素及其衍生物的合理利用,也有利于基于机制协同的联合应用,降低青蒿素类化合物的耐药问题,同时也会促进类似机制抗疟新药的筛选和研发。众所周知,青蒿素类化合物的激活主要依赖于血红素或者亚铁,产生活性氧及其他自由基。 近几年,在肿瘤学研究中发现,青蒿素及其衍生物能诱导肿瘤细胞铁死亡(ferroptosis),青蒿素及其衍生物是铁死亡的诱导剂;但是,其抗疟机制中是否有铁死亡的参与,尚未见报导。铁死亡是近年新发现的一种调节性细胞死亡模式,在形态学、基因学、生物化学特征方面与传统凋亡、坏死、自噬等死亡模式存在显著差异,本质表现为Fe2+依赖的脂质过氧化物超限蓄积导致的细胞质膜损伤。 由于疟原虫与肿瘤细胞等在生物学上明显不同,铁死亡是否为恶性疟原虫的
·他汀类非降脂作用专题· 他汀类药物的抗血栓作用 黄全跃 赵水平 (中南大学湘雅二医院心内科,湖南 长沙 410011) [关键词] 血栓形成/药物疗法; 降血脂药 [中图分类号] R972.6 [文献标识码] C [文章编号] 1671-7171(2004)01-0012-03 他汀类药物在冠心病(CHD)的一级预防和二级 预防中均获有显著效果。他汀类对心、脑血管的保 护作用看起来要比所期待的降脂效应大,提示存在 有降脂以外的作用。近年来越来越多的动物实验和 临床证据表明,他汀类对血小板和凝血/纤溶系统有 明显的影响,具有良好的抗血栓作用。 1 对血小板的影响 业已证明,血小板聚集在动脉血栓形成过程中 有着启动和强化作用。高胆固醇血症患者的血小板 比正常胆固醇对照者的血小板对聚集剂的促聚作用 更加敏感。有研究发现ADP诱导的纤维蛋白原与 血小板的结合是由低密度脂蛋白(LDL)而介导,且 呈现LDL剂量依赖性[1]。 有研究者观察到,他汀类能明显抑制血小板的 聚集作用。可能的机理有多种:有人认为,他汀类一 方面通过降低体内胆固醇的水平;另一方面改变血 小板膜的胆固醇含量从而影响膜的流动性,但是这 些似乎均与他汀类的降脂作用有关[1]。近年来的体内、外研究显示,内皮衍生的一氧化氮(NO)除了调 节血压、增加局部血流外,还能减轻白细胞的激活和 抑制血小板聚集。而他汀类则间接通过增加内皮NO的产生和生物利用来抑制血小板的聚集,不依 赖于血浆胆固醇水平。动物实验显示[2],阿托伐他 汀能上调正常胆固醇小鼠血小板内皮Ⅲ型一氧化氮 合成酶(eNOS)的mRNA表达水平和降低血小板激 [作者简介] 黄全跃(1960-),女,湖南邵阳人,心内科教授,主要从事血栓与动脉粥样硬化的研究。 [收稿日期] 2003-11-06活而不降低胆固醇水平,其中血小板激活的指标血小板4因子(PF4)和β-血栓球蛋白(β-TG)显著下调且有剂量依赖性。有趣的是,在eNOS敲除的小鼠,阿托伐他汀不能影响其PF4和β-TG的水平。研究证实,激活的血小板释放的NO可显著抑制血小板募集因而限制血管内血栓的进展。该项研究提示,他汀类治疗确实主要是通过上调eNOS而起抗血小板激活作用的。特别值得一提的是,最近Gertz 等[3]报道,阿托伐他汀治疗14d使急性脑梗死小鼠的PF4和β-TG的水平显著下降,但是停药2d以后,两者的水平分别上调2.9和3.1倍;同时阿托伐他汀治疗使主动脉和脑血管的eNOS上调2.3和1. 7倍,但同样仅在停药2d以后,上述二处的eNOS 分别下调5倍和3.1倍,由此说明急性撤退他汀类药物,2d后即丧失对小鼠脑缺血和血栓形成的保护功能。 2 对凝血功能的影响 动脉血栓形成的过程是血小板黏附、聚集、释放和瀑布式的血液凝固的过程。血液中的多种凝血因子之间的活性失衡及过度激活是血栓形成的重要条件。不少文献报道,急性冠脉综合征(ACS)或急性脑梗死时部分凝血因子水平或活性发生改变。 他汀类除了抗血小板的作用以外,还通过影响凝血系统发挥着抗血栓形成的功能。组织因子(TF)是表面结合蛋白,存在于巨噬细胞、内皮细胞和人动脉粥样硬化斑块的平滑肌细胞,是外源性凝血系统的启动因子。TF介导的凝血过程受组织因子途径抑制物(TFPI)的抗衡。研究显示[4],他汀类药物可减少人类培养单核细胞/巨噬细胞(氟伐他汀
青蒿主要药用成分青蒿素的衍生物是目前疗效最好 抗药性最低 Newly compiled on November 23, 2020
广西青蒿(黄花蒿)产业发展规划 广西壮族自治区农业厅 二○○年五月
目录
青蒿主要药用成分青蒿素的衍生物是目前疗效最好、抗药性最低、应用前景最好的抗疟药物,而且青蒿素在深度开发方面也有很好的市场前景。广西是全国2个青蒿素产品主要产地之一,随着全球市场对青蒿素的需求量不断扩大,青蒿产业面临良好的发展机遇。抓住机遇,加大工作力度,把广西壮族自治区建设成为青蒿生产基地的意义非常重大。按照自治区主要领导同志提出的“把广西建设成为青蒿生产基地”指示,充分发挥广西壮族自治区优势和特色,把握机遇,加快发展,培植国民经济新的增长点,探索中草药现代化产业开发新途径,制订本规划。 一、青蒿产业的现状及发展前景 (一)WHO改变用药配方,青蒿素需求强劲 据世界卫生组织(WHO)统计报告,全世界每年急性疟疾患者达3亿人,每年死于该病的人数约200-300万,90%的死亡病人发生在非洲,其中5岁以下儿童超过90%。曾是抗疟疾特效药的奎宁,长期使用后会产生广泛抗药性。而青蒿素类药物经多方试验,证明其在抗氯喹原虫耐药株恶性疟等方面有特殊疗效,1990年在越南疟疾患区使用,治愈率达97%,受到患区当地政府和患者的普遍欢迎。 2001年12月中旬, WHO的一份公报指出“治疗疟疾的最大希望来自中国”,肯定青蒿类药物为治疗疟疾的“首选药物”。2004年2月,WHO确定将青蒿琥酯、蒿
甲醚等青蒿素类药物作为全球新一代抗疟药,同时针对青蒿类药物半衰期短,治疗期较长(7天),价格较高的问题,推荐治疗期较短(3天)、相对便宜的以青蒿类药物为基础的联合用药(简称ACTs疗法),逐步取代传统的治疗疟疾方案。目前全球已有40个国家选择了ACT作为官方治疗疟疾用药,其中有36个国家用其作为一线治疗药物,4个国家作为二线药物,其它还有14个以上国家最近正考虑改换成ACT药物。据WHO统计,2003年全球抗疟药销售额约15亿美元,青蒿类药物销售额约为其1%,青蒿类药物市场空间巨大,发展前景看好。 2004年11月16日,由联合国儿童基金会、WHO、全球基金在哥本哈根召开了“全球抗疟药供应商预认证”会议。在这次会议上正式公布了联合用药的推荐处方及会议主持者的采购量,WHO对2004年的联合用药(ACTs)政策做了调整:确定了4个处方作为替代奎宁类的抗疟推荐用药,进入公共采购目录的药物主要包括青蒿琥酯和蒿甲醚等,以青蒿琥酯为主的联合用药2005年国际组织、机构的部分采购金额(不包括疟疾区各国政府的采购)共计4000万美元,2006年为4800万美元。估计各国政府采购金额、商业公司采购金额不会低于国际组织、机构的采购金额。目前青蒿素用药缺口为每年总需求的40~60%,估计填补全球青蒿(黄花蒿)种植的空缺需要15~20年。
他汀类药物的合理应用 血脂异常,包括低密度脂蛋白(low-density lipoprotein, LDL)升高和高密度脂蛋白(high-density lipoprotein, HDL)降低是冠心病(Coronary Heart Disease,CHD)的一个重要危险因素[1],降低LDL是治疗CHD的最有效措施之一 [2]。血脂异常的治疗包括治疗性生活方式改变、药物治疗、血浆净化疗法、外科治疗及基因治疗。药物治疗是最常被使用的方法,其中又以他汀类使用最广泛,已有充分的证据表明他汀类药物是降低动脉粥样硬化性心脏病高危患者LDL水平的一线用药[3、4]。本文就当前他汀类药物应用中的一些问题做一综述。 一、回顾 他汀类药物是一类被广泛应用的降胆固醇药物,通过作用于肝细胞3-羟基-3-甲基戊二酰辅酶A(HMG-CoA)还原酶(催化胆固醇生物合成中的限速步骤),抑制胆固醇合成,并上调LDL受体,加速清除循环中的LDL颗粒,从而降低血浆LDL水平。 [Grundy SM. HMG-CoA reductase inhibitors for treatment of hypercholesterolemia. N Engl J Med, 1988,319:24-33]自从1987年首获批准后,特别是上世纪90年代5项具有里程碑意义的大规模临床试验发表以来,诸多以他汀类作为治疗组进行的高质量的随机试验已经明确表明:他汀类药物具有降低血管性死亡、非致死性心肌梗死、卒中以及动脉血运重建风险的能力,[Cholesterol Treatment Trialists’(CTT) Collaborators. Efficacy and safety of cholesterol-lowing treatment: prospective meta-analysis of data from 90056 participants in 14 randomised trials of statins. Lancet,2005,366:1267-78] 并为有关指南的制定、修改提供了充分的科学依据。 循证医学证明了他汀类降脂治疗的临床益处,降低LDL 30%-40%、使CHD的发病率降低24%-36%,并明确了他汀类降脂药物在推荐剂量下长期应用具有良好的耐受性和安全性。综观2004年NCEP ATPⅢ的最新报告、欧洲心脏病学会(ESC)及美国ADA指南可以看到,尽管因各国家和地区的具体情况不同而使其各自的标准有所差异,但都强调降脂治疗的首要目标是降低LDL水平。当前全球降脂趋向是:针对高危人群;首选他汀类降脂药;LDL水平“越低越好”。有证据显示:无论受试者LDL的基础水平如何(即
他汀类药物治疗高胆固醇的药理作用 大量的临床资料表明,高脂血症是动脉粥样硬化或冠心病的主要危险因素,降脂可明显减少心脏事件的发生,降低心血管发病率及总死亡率。他汀类药物已被广泛用于临床,该药不但有显著的降脂作用,而且具有改善血管内皮功能,稳定动脉粥样硬化斑块,保护和修复血小板膜,抗骨质疏松,抑制心肌成纤维细胞增殖和胶原合成,几乎可以干预动脉粥样硬化的各个环节,明显促进斑块回缩,从而对防治心脑血管疾病事件的发生起到积极作用。 1 他汀类药物的理化特性及其作用 他汀类药属于3一羟基3一甲基戊二酰辅酶A (.该类药通过竞争性抑制肝脏合成胆固醇的限速酶HM(}Ⅸ)A还原酶的活性,使肝内胆固醇合成减少,触发肝脏高密度脂蛋白受体的表达增加,从而使循环中有更多的高密度脂蛋白及其前体进入肝脏与 LDH受体结合而被清除。他汀类药具有高效降高密度脂蛋白和中效升砌iC作用,降rig作用的强度则与TG基线水平及降高密度脂蛋白强度有关。 目前,临床应用他汀类药物有:辛伐他汀、洛伐他汀、普伐他汀、氟伐他汀、阿托伐他汀和西力伐他汀,前三种为天然药物,后三种为人工合成药物。他汀类药物吸收不佳,口服给药不到5%可进入循环系统且需经过肝脏,因此循环水平较低。他汀类药物对缺血性心脏病的长期干预(L I Pm)与冠心病事件复发研究(C』6衄) 显示此类药物不仅能降低低密度脂蛋白一胆固醇,还具有稳定动脉粥样硬化斑块、改善内皮功能、抗炎、抑制血小板聚集及改善胰岛素抵抗等。 2降脂作用 2.1抑制平滑肌细胞(VSMC)增殖、迁移平滑肌细胞的增殖、迁移是动脉粥样硬化发病过程的一个重要环节,在动脉粥样硬化的形成过程中,平滑肌细胞由收缩变成合成型,并迁移至内膜下。合成型的特点是:类似纤维母细胞,含少量肌丝而含大量粗面内质网及高尔基体,可以分泌许多基质加入斑块中。平滑肌细胞本身还可转变成泡沫细胞。他汀类药物的抗动脉粥样硬化作用可能部分是通过对细胞的直接作用介导的。Planavila等发现,阿托伐他汀能抑制NF-kB活化,提高过氧化物酶体增殖物活化受体 (PPAR)水平,减少PPAR蛋白与NF—kB p65亚单位的结合,从而阻断NF-kB信号转导途径,延缓心肌肥大。这种作用与其抑制羟甲基戊二酰辅酶A (HMG-CoA)还原酶,使甲羟戊酸衍生物的产生减少有关。他汀类可下调人及动物VSMG细胞周期依赖蛋白激酶2表达,诱导原癌基因p53,上调细胞周期依赖蛋白激酶抑制剂表达。 2.2改善血管内皮功能高胆固醇血症、高血压等可造成内皮结构与功能损害,促使血管损伤。研究证明,洛伐他汀可在治疗高胆固醇血症的同时具有明显改善内皮功能、改善微循环、降低胰岛素抵抗作用。45名冠心病高胆固醇血症患者服用洛伐他汀治疗8周,其结果治疗后4周较治疗前内皮素(ET) 血栓素、甲襞微循环(NFM)总积分值显著下降,6一酮一前列腺素、一氧化碳、胰岛素敏感性(IS)、指数明显升高;治疗后8周与治疗前比较TXB2、NFM总积分值进一步下降(P均d0.01)。研究证明,洛伐他汀能够降低内皮素改善血管内皮功能,扩张小动脉改善微循环增加骨骼肌血量,提高胰岛素受体。在动物实验中,他汀类药物能减少心力衰竭大鼠血小板的活化,改善内皮功能。阿托伐他汀治疗4个月能逆转内皮功能紊乱,延长治疗时间还可发挥抗炎作用。上述实验表明,
中国科学: 生命科学 2012年 第42卷 第5期: 345 ~ 354 SCIENTIA SINICA Vitae https://www.doczj.com/doc/74718437.html, https://www.doczj.com/doc/74718437.html, 英文引用格式: Sun C, Li J, Zhou B. Mechanism of action of artemisinins: a long unsettled challenge. SCIENTIA SINICA Vitae, 2012, 42: 345–354, doi: 10.1360/052012-168 《中国科学》杂志社 SCIENCE CHINA PRESS 评 述 青蒿素类药物的作用机制: 一个长久未决的基础 研究挑战 孙辰, 李坚, 周兵* 清华大学生命科学学院, 生物膜与膜生物国家重点实验室, 北京 100084 * 联系人, E-mail: zhoubing@https://www.doczj.com/doc/74718437.html, 收稿日期: 2012-04-20; 接受日期: 2012-04-24 doi: 10.1360/052012-168 摘要 青蒿素是中国自主研制的抗疟良药, 高效、低毒, 许多基于青蒿素研发的衍生物具有良好的抗疟效果, 近年来已成为抗疟的一线药物, 受到世界医疗卫生界的充分肯定. 虽然青蒿素结构奇特, 抑疟效果显著, 但40年来其生物作用机制之谜一直未被彻底破解. 针对青蒿素类药物的作用机制, 提出了不同的假说, 如血红素参与青蒿素的激活并被烷基化从而起到抑疟作用, 线粒体参与青蒿素的激活和作用过程, 某些特定的蛋白是青蒿素作用靶点等. 除抑疟外, 青蒿素类药物在杀灭其他种类寄生虫、抑制某些癌症细胞以及抗病毒、治疗类风湿等方面也有一定作用. 本文将对青蒿素类药物作用机制的研究进行综述及展望, 包括抗疟疾过程中的药物激活、作用靶点以及简要的青蒿素抑制肿瘤细胞作用机制, 以期为今后的研究提供帮助. 关键词 青蒿素 作用机制 血红素 线粒体 2011年9月, 中国中医科学院科学家屠呦呦获得美国拉斯克临床医学奖, 以表彰她在抗疟药物青蒿素(artemisinin)开发过程中的贡献. 一时间, 疟疾和抗疟良药青蒿素成为国人关注的焦点. 疟疾是严重影响人体健康的传染性疾病, 尤其在非洲、南亚、东南亚及南美洲大陆的热带亚热带地区发病严重. 世界卫生组织统计报告显示, 疟疾2010年发病人数为2.16亿, 造成约65.5万人死亡, 其中86%的受害者是5岁以下儿童[1]. 青蒿素的发现是中国20世纪70年代“中国疟疾防治药物研究工作协作项目”(又称“523”科研项目)中的一项重要成果, 中国科学家们的协作研发为世界人民抗击疟疾做出了重要贡献. 青蒿素对红内期疟 原虫有直接杀灭作用, 快速高效且毒性低, 缺点是半衰期比较短, 单独使用再燃率较高. 其他治疗疟疾的药物, 如氯喹、甲氟喹和奎宁等均出现了抗药株, 而青蒿素类药物自投入使用以来, 除在柬埔寨地区出现青蒿琥酯对疟原虫清除效率降低的病例(体外实验并没有出现类似的延缓现象)[2], 尚无确证的青蒿素类药物抗性疟原虫出现. 为防止将来青蒿素抗性菌株的出现, 世界卫生组织颁布了 “青蒿素联合疗法”(artemisinin combination therapy, ACT). 除治疗疟疾的良好效果外, 青蒿素在杀灭其他种类寄生虫, 抑制某些癌症细胞以及抗病毒、治疗类风湿等方面也有一定作用. 青蒿素的治疗效果举世瞩目, 但其作用机制至今仍是一个谜, 也是一个长期