
第六章 脂肪族亲核取代反应
R L +Nu
-R Nu +L -
底物 亲核试剂 离去基
6.1试剂与亲核性
具有亲核性的试剂叫亲核试剂,亲核性是指试剂与正电性碳原子的成键能力,亲核试剂可以是中性分子和离子。不同亲核试剂的亲核性不同,亲核试剂均为路易斯碱,而碱性(质子理论)是指试剂与质子的结合能力,多些情况下二者的强度一致。 1、 亲核原子相同时,亲核性大小与碱性一致。
R O ->H O ->Ar O ->RCO O ->R O H>H 2O
CH 3
O -O -O -
Cl NO 2
O -
>>
>
取代基的供电性越强,亲核性和碱性越强。
2、 亲核性原子为同周期元素的原子时,其亲核性与碱性大致一致。
R 3C > R 2N - >RO - > F -
3、 同族元素原子的亲核性大小与碱性相反,即原子半径较小者,碱性较大,而亲核性则较小。 PhSe ->PhS ->PhO - I ->Br ->Cl ->F -(溶剂化最强)
亲核性 亲核性(这是质子性溶剂中) 4、 空间效应
碱性 CH 3O -<(CH 3)3CO - 亲核性 CH 3O ->(CH 3)3CO -
N
R +CH 3I N
CH 3
R I -
亲核性
N
CH 3
N
N
CH 3N
CH 3
CH 3
N
C(CH 3)3
>
>
>
>
相对速度
2.3 1.0 0.5 0.04 0.0002
空阻
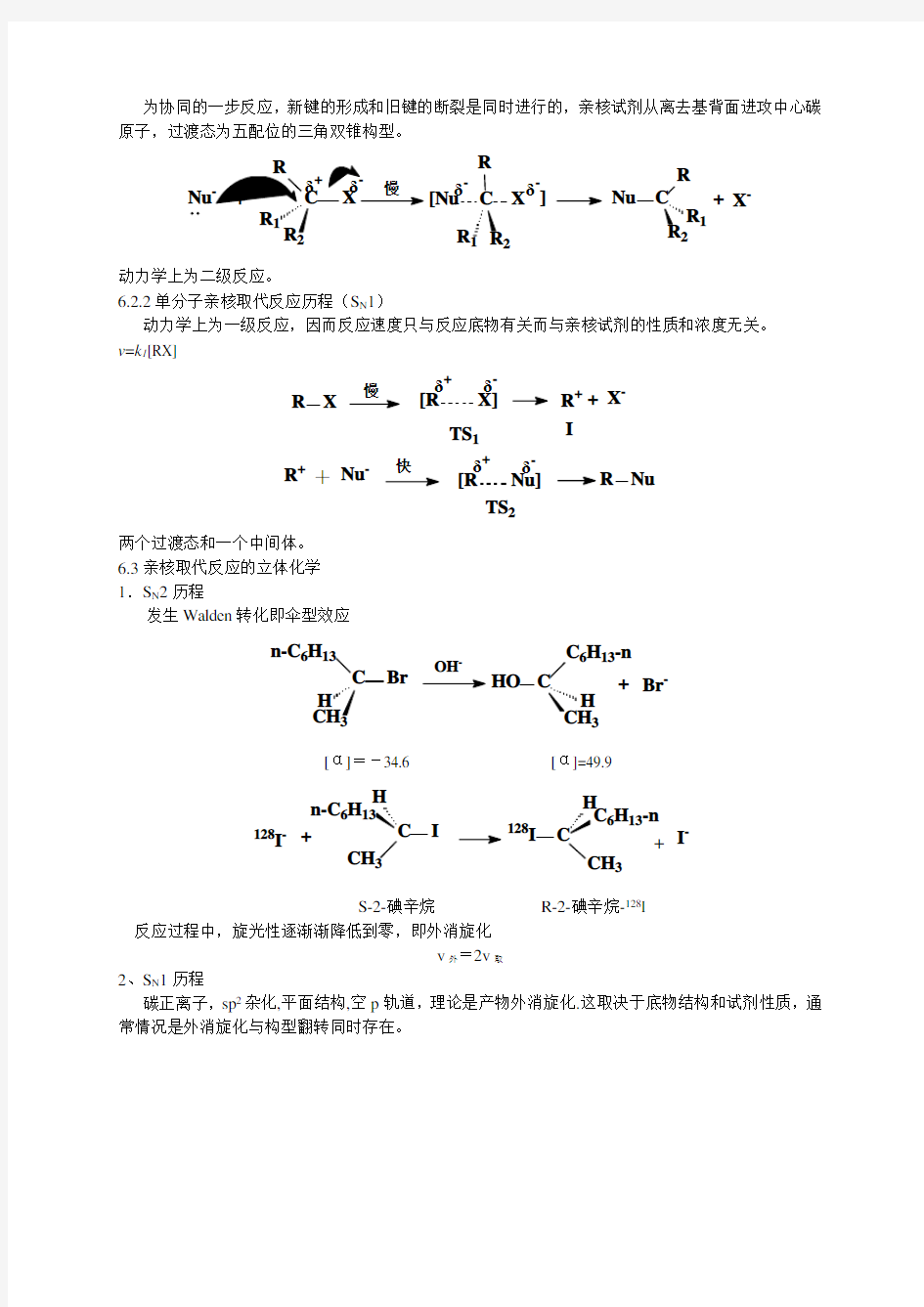
6.2亲核取代反应历程(S N 2)
为协同的一步反应,新键的形成和旧键的断裂是同时进行的,亲核试剂从离去基背面进攻中心碳原子,过渡态为五配位的三角双锥构型。
Nu
C [Nu C X R
2
δ-]Nu
C R
R 1
2+X -δ-+
动力学上为二级反应。
6.2.2单分子亲核取代反应历程(S N 1)
动力学上为一级反应,因而反应速度只与反应底物有关而与亲核试剂的性质和浓度无关。 v =k 1[RX]
R +
+Nu
-
[R Nu]δ+δ-R Nu
快
R X
[R X]R ++X
-δ+δ-TS 1
TS 2
I
两个过渡态和一个中间体。 6.3亲核取代反应的立体化学 1.S N 2历程
发生Walden 转化即伞型效应
C n-C 6H 13
Br
OH -
HO C
C 6H 13-n
H 3
+Br -
[α]=-34.6 [α]=49.9
+I
-128I
-+C n-C 6H CH 3
I
128
I
C 6H 13-n
CH 3
S-2-碘辛烷 R-2-碘辛烷-128I
反应过程中,旋光性逐渐渐降低到零,即外消旋化
v 外=2v 取
2、S N 1历程
碳正离子,sp 2杂化,平面结构,空p 轨道,理论是产物外消旋化.这取决于底物结构和试剂性质,通常情况是外消旋化与构型翻转同时存在。
C R 2
Nu 1
2Nu
+50%
50%
当形成的碳正离子稳定性较小时,当被试剂进攻时,此时离去基尚未离开,在一定的程度上产生了遮蔽效应,因而妨碍了Nu
-从离去基的方向进攻,故主要得构型反转的产物;如果试剂浓度低,碳正离子可顺利成为自由离子,和离去基X -分开后才接受Nu -进攻,这时产物外消旋化。如果进攻试
剂的浓度较高,被X -遮蔽的碳正离子受试剂进攻的机会增加构型反转比例增加,在S
N 1反应中,还有一些产物不是从离域的碳正离子中间体形成的,而是有离子对形成的。 离子对历程
R
X
R +X
-
R +X -R ++X -
-S N 2
构型翻转
紧密离子对
构型翻转
自由碳正离子
外消旋化(100%)构型翻转 > 构型保持
(部分外消旋化的同时,有部分构型翻转)溶剂分离对
-
-
Nu -N 1
底物
6.4邻基参与
如果亲核试剂与离去基共处于同一分子中,则发生分子内S N 2。
HO CH 2CH 2Cl
Ca(OH)2
CH 2CH 2
Cl -CH 2
CH 2
O
ClCH 2CH 2CH 2CH 2NH 2
N H
KOH 2
离去基Cl 的离去是由氧负离子(或氮原子)的帮助下完成的,通常称为邻基协助,邻基参与或邻基效应(分子内的S N 2)
条件:当邻近基团具有未共用电子对时,在与反应中心的距离适当时,就发生邻基参与。 结果:①生成环状化合物,②限制产物的结构,③反应速度异常增大。 6.4.1 n 参与 1.氧参与 包括:
-3O
O CHO O ,
,
,
,
,
,
MeO OTs CH COO -
Me OAc
k 反/k 顺=6 反式的产物构型保持不变
MeO
OAc
-
H H OAcOTs
H OAc H
OAc
CH 3COO -CH 3COOH
k 1=2.9×10-7L.mol -1.S -1
这是一个S N 2历程,由于AcO 的吸电子作用使得碳正离子的形成较困难。
OAc H
H
OTs
OAc H
H
OAc CH 3COO -3+
H
OAcH
OAc
外消旋体
k 2=1.9×10
-4
L.mol -1.S
-1
+
Me
OAc H H
OAc H OAc OAc
H O
C Me
50%
50%
镜面对
称性(非手性分子)
只有一个手性碳且反应中心为手性碳时,邻基参与的结果将是构型保持。
C Et
Et
HO C -
C Et Et
-
O
Cl
-Cl -
C Et
O
Me
C
Et
Et -
O C OH 1
2
3
1变成2为邻基参与(构型翻转),2变成3为S N 2反应(构型翻转,3的构型和1与底物相同)。因此邻基参与和S N 2反应的总结果为构型保持。
C Br O C
O C C
HO O -O
总结果为构型保持 2.硫参与
ClCH 2CH 2S
CH 2
22CH 2
CH CH 2
-
S N 2
ClCH 2CH 2CH 2
CH 2OH 邻基参与
反应速度较快
3.卤素参与
CH 3
H OH Br H
CH 3
H
CH 3
OH Br CH 3
H
H CH 3
Br Br CH 3
H
赤式dl 对
赤式meso
+
H
CH 3OH Br H
CH 3H CH 3
Br Br H
CH 3
CH 3
H
OH Br CH 3H
CH 3H
Br Br CH 3
H
HBr
++
苏式dl 对
苏式
dl 对
H CH 3Br
Br H
CH 3CH 3
H
Br Br CH 3
H
+
Br H CH 3H
CH 3
OH 3H +
H CH 3
-H 2O
(2S,3S)
(2S,3S)
(2R,3S)
(2R,3R)
(2S,3S)
对称性
23
CH (2R,3S)
(2S,3S)
H Br H
Br
CH 3
CH 3meso
CH H 3
6.4.2 π参与 1.C=C 的参与
AcO -
构型不变
k 乙酸解
1011
1
k 乙酸解
113.2H 3H 3148
π供电性增加,速度增加。C O
NO 2
NB :
2.苯基参与
苏式(2R,3S)
进攻C
进攻C3
C
H C C
3Ph
OAc
C
Ph
CH 3
苏式dl
(2S,3R)meso
(2R,3S)
(2S,3R)
50%50%
50%构型转化,50%构型翻转,结果为外消旋化。
赤式(2R,3R)进攻C
进攻C3
C
CH
C C
3Ph
OAc
3
赤式(2S,3S)(2R,3R)
-
CH
手性
(2R,3R)
构型保持不变。
6.5影响亲核取代反应的因素 6.5.1烃基的结构 1.对S N 1影响
凡是能稳定C +的各种因素均有利于S N 1的进攻,中心碳原子上连接的供电基团,能分散碳正离子上的正电荷,提高碳正离子的稳定性。 (1)电子因素
稳定性:3°R + > 2°R + > 1°R + 烷基的+I 和+C ’ S N 1的速度:3°RX > 2°RX > 1°RX (2)空间因素
C X
R 慢
sp 2
张力得到了释放sp
3
R'
(3)苯基或乙烯基取代的卤甲烷按S N 1进行
R Cl +CH 3CH 2OH
S 1
R OC 2H 5+HCl
(4)当杂原子如O,N,S 等原子直接与中心碳相连时,按S N 1进行
C 2H 5O CH 2Cl CH 3CH 2CH 2CH 2
Cl C 2H 5O CH 2CH 2Cl
k 相对
109
1
0.2
+C>-I ,有利于碳正离子的形成
杂原子上的孤对电子与中心碳的空p 轨道交盖形成离域π键,使碳正离子稳定。 (5)卤原子位于桥头碳上时,只能按S N 1进行,但反应速度极慢。
25℃,80% H 2O-C 2H 5OH
k 相对
Me 3CBr
Br
1
10-3
10-7
10-13
桥头碳难于形成平面碳正离子,刚性越大(桥原子数减小),反应速度越小。 2.S N 2的影响
主要影响因素为空间效应
Nu -+
C R
X
慢
(Nu C X R δ-δ-)Nu
C R
R''+X -
过渡态为五配位,张力增大
因此不论是α-C 还是β-C 上的取代基增多均不利于S N 2反应。
Nu
-
S N 2
Nu
R +X -
R X +
H 33C
3
空阻很大
6.5.2离去基的影响
不论S N 1还是S N 2的定速步骤中均存在C-X 断裂,故C-X 愈易断裂,反应速度愈快。 稳定性 F -
OH -R OH +Br -
R
Br +OH -为不好的离去基
H +
R OH
R OH 2
Br -
R
Br +H 2O
H 2O 为好的离去基
6.5.3试剂的影响
试剂的亲核性对S N 1几乎无影响,对S N 2影响较大。 (1)亲核性越大,越有利于S N 2;
(2)亲核试剂的体积越小越有利于S N 2;
(3)强亲核试剂导致产物构型反转,故弱亲核试剂导致产物部分外消旋化。 6.5.4溶剂的影响
1.对S N 1反应,增加溶剂极性和溶剂的离子—溶剂化能力,导致反应速度显著增大。
(1)在定速步骤中,反应物电离成过渡态所需的大部分能量可由在溶剂和极性过渡态之间形成偶极-偶极键来供给。溶剂的极性越大,则溶剂化能力越强,电离作用越快。
(2)碳正离子或定速步骤的过渡态极性大于反应物,前者因溶剂化而稳定,极性溶剂有利于S N 1,且随着溶剂极性增强而加速。 2.对于S N 2
L -Nu -
+R L
(Nu R L)Nu
R +δ+
δ-电荷分离
溶剂极性增大,不利于反应;极性减小,有利于反应。
L -Nu +R L
(Nu R L)
Nu
R +δ+δ-极性增加
溶剂极性增大,有利于反应。
极性降低
Nu -L
+R L +
(Nu R L)
Nu R +δ-δ+
溶剂极性增大,不利于反应
3.极性质子性溶剂使亲核试剂溶剂化增加亲核试剂的稳定性,降低其亲核活性。
NuR RO H
Nu -
H H OR OR
△G'3ROH Nu -
+(Nu
R L)
+δ-δ-R L △G''
L
△G'+△G''=△G
这不利于S N 2
4.极性非质子溶剂如DMF,DMSO,HMPT 等,这时亲核试剂完全暴露未溶剂化,对S N 2有利的。
CH 3I +NaN 3
N CH 3N 3
+
NaI
6.6 具有双亲核中心的两极性的反应方向 两可性试剂如
O N=O C CH
1.发生S N 1时,电负性大,电子云密度高的原子将发生亲核取代。
R Br +Ag +C N C +AgBr 得到异腈
R Br
R N C
Ag +
R
+
C
N
电子云密度大
-
2.在S N 2中,电负性小的原子为亲核中心。
+R Br
S N 2
(N C R Br)
δ-δ-N C R +Br -
C N
Na +
这时电负性较小的原子周围溶剂化作用较弱,去溶剂化所需能量小,其供电性强,有较强的极化作用,故其亲核性较强。 3.底物烷基结构的影响
R X +O
N
R O N O
R
O -O
+
亚硝酸酯
硝基烷
亚硝酸根
卤烷产率
亚硝酸酯
硝基烷
1°RX
2°RX 3°RX 降低(空阻增大)
故烷基体积增大时,有利于S N 1反应,烷基体积越小,越有利于S N 2。
6.7亲核取代反应在有机合成中的应用 6.
7.1卤烷的亲核取代反应
可发生官能团的转化或碳链的增长 1. 卤烷的水解
CH 2=CHCH 2CH 3
CH 2=CHCHCH 3
Br
CH 2=CH CHCH 3
OH
2NBS
竞争使反应有消除反应(浓度和强碱及烯丙式醇3°RX 特别易发生E)
CH 3CH 2C CH 3
Br CH 3
+H 2O
NaOH
CH 3CH 2C CH 3
OH CH 3+CH 3CH 2=C
CH 3
CH 3+CH 3CH 2C CH 2CH 3
1%
99%
易发
生消除反应的3°RX 宜采用碱性较弱的醋酸盐形成乙酸酯,再水解得醇。
CH 3CH 2C CH 3
Br CH 3
+3CH 2C CH 3O CH 3
C O
CH CH 3COOAg
2CH 3CH 2C CH 3
OH
CH 3>90%
2.williamson 反应
CH 3CH 2ONa +Br
CCH 2CH 3CH 3
CH 3E
CH 3CH CMe 2
CH 3CH 2Br +CH 3CH 2C CH 3
ONa
CH 3
CH 3CH 2OC CH 3
CH 2CH 3
CH 3
N
3.卤烷的氨解
一般得混合物,如果NH 3大大过量,1°RNH 2将为主要产物,要获得纯伯胺用gabriel 法。
C
C N -O O
+RX COOH
COOH
C C NR O O H O +
+RNH 2
4.卤烷与有机膦的反应
RCH 2X +PPh 3
RCH 2PPh 3X -n
RCHPPh 3
Wittig 试剂
5. 与碳亲核试剂的取代反应 使碳链增长
(1) 氨解形成多一个碳的腈及羧酸等
RX +NaCN
乙酸
H 2O
RCN
3+R COOH
(2)炔化反应
n BuBr +NaC CH n BuC CH
Na n BuC C Bu n
合成末端炔和非末端炔
(3)偶联反应(与烷基铜锂的反应)
CH 3Br +(CH 2
C)2CuLi
CH 3
3
C CH 3CH 2+CH 2=CCu CH 3
+LiCl
Cl +CH 3(CH 2)3Cl
CH 2CH 2CH 2CH 3
Wurtz-Fittig 反应
(4)与含活泼亚甲基类化合物的取代反应。
首先与强碱作用,形成碳负离子亲核试剂,然后再 -C 上引入烷基或烃基
CH 3CCH 2COOC 2H 5
O
EtONa RX
CH 3CCHCO 2
OR ①②EtONa R'X ①OH -+2
2
CH 3CCH 2
R O RCH 2COOH
CH 3CCCO 2Et
O CH 3
CH
3
酮式分解
酸式分解
CH 3CCHRR'O
CHCOOH R
R'
与二元卤烷可分别形成二元甲基酮,二元酸或环烷基甲基酮或环烷基乙酸。 6.7.2醇的亲核取代反应
由于-OH 为不好的离去基,其亲核取代反应一般要在酸性催化下进行,这时H 2O 是一个好的离去基,另外可转化一个磺酸基离去基。
n Pr 2C CHCH 2OH +CH 3SO 2Cl
n Pr 2C CHCH 2OSO 2CH 3n Pr 2C CHCH 2Cl
6.8羧酸及其衍生物的亲核取代反应 6.8.1反应历程与影响因素 1、反应历程
过程为加成-消除历程,结果为亲核取代。与S N 2是不一样(分两步进行)。 亲核加成
-C
L
Nu
C R O -L
消除
Nu
C R O -L
Nu
C R
O +L - 酯的碱性水解
HO -+R
C OR'
O*
R C OH
-OR'R C OR'
O -*
OH
R C OR'O
+HO*-
这样水解后酯中18
O 含量大大减小,故非一般的S N 2,如为S N 2反应,则酯的18
O 含量不变。 2、结构与活性的关系
+C
R
-I, -I 有利于亲核加成,+C 不利于亲核加成。
L -X OCR
O
OR NH 2
-I 减小+C
增大
L 离去能力
减小
反应活性
O
>O O
>RCOOR >RCONH 2
6.8.2
酯化反应
RCOOH +R'OH
+
RCOOR'+H 2O
1、Ac2(双分子酰氧断裂) 伯醇的酯化通常为此历程
R C OH
OH HOR'
C 2
OH
OR'
H 2O
R C OH OR'
R C OR'O
+H +
R C OH O 快
R C OH OH R C
OH
OH
2、A A11(酸酯消化下的烷氧断裂) 3°醇的酯化就是此历程
R''COOH
2-H +
R''COCH 2RR'R''
O
次
R'CH
CRR'
主
R'CH 2C R
OH
R''
+
R'CH 2C R
R''
R'CH 2C
R''
R
亲核性H 2O>羧酸;故脱氢离子以烯为主要产物 6.8.3间接酯化-羧酸衍生物的醇解反应
酰卤和酸酐的间接酯化可避免直接酯化的可逆性,而且可以同叔醇酯化。
(CH 3)2CHCCl O
+Me 3PhNMe 干醚 回流
Me 2CHCOCMe 3
O
(CH 3CO)2O +
COOH
OH
H +
△
COOH
OCCH 3
O
酯交换反应
RCOR'O
+R''OH
H +-
RCOOR''+R'OH
RCOR'O
+CH 3COOH
+
CH 3COOR'+RCOOH
要求R ’O -碱性比R ’’O -弱,离去能力R ’O ->R ’’O -,而CH 3O -的碱性最弱,故常用甲醇的酯作为反应底物
CH 2=CHCOCH 3O
+(CH 3)2CHCH 2CHOH
CH 3
CH 2=CHCO CHCH 2CHMe 2
CH 3O
Y95%
蒸去CH 3OH
Me 3COC
COCMe 3
O
O CH 3O C
C OCH 3O
O
+Me 3
COH
380 C
6.8.4羧酸衍生物的氨解 1.羧酸的氨解
RCOOH +R'NH 2(过)
可逆
RCNHR'O
+H 2O
除去水用PCl 3,TiCl 4等
2PhCOOH +6Et 2NH
TiCl 2PhCONEt 2O
+4Et 2NH
2.羧酸衍生物的氨解
PhCCl O
+H
2N CHCOOH
Ph
PhCNHCHCOOH
O
Ph
+NH 3
NCCH 2CNH 2O
+C 2H 5OH
NCCH 2COOC 2H 5
CN -吸电基有利于取代
CH 3CH 2COOC 2H 5+PhNH 2
CH 3COONa 苯、回流
CH 3CH 2CNHC 6H 5
弱碱
6.8.5晴的溶剂解
R C N +H 2
O
H +
(R C NH)
OH RCNH 2
O H 3+O
RCOOH
控制水解可用酰胺
PhCHCN +H 2O
+
PhCH 2CNH 2
O
45 C
室温下,晴可用HCl-HCOOH 溶剂水解为酰胺,而其它可水解的基团如OR -,Cl -,Br -等不受影响。
Cl
CN +H 2O
HCl HCOOH
Cl
CNH 2
O
RC N +HCl(g)+R'OH
R C
OR'
NH.HCl H O
RCOOR'+NH 4Cl RC OR''
OR''OR''原羧酸酯C NH 2.HCl NH
脒
教学目标:掌握各种因素对亲核取代反应机理的影响。 教学重点:烷基结构、亲核试剂、溶剂等因素对S N1 和S N2 反应的影响 教学安排:H 1,H3 —>H4;40min 基本概念:溶剂解:溶剂作为亲核试剂的亲核取代反应,称为溶剂解或溶剂解反应。溶剂解反应可根据所用的溶剂是水、乙醇还是乙酸,分别称为水解、乙醇解,乙酸解等。 卤代烷的亲核取代反应,既可按S N2 亦可按S N1 机理进行,但究竟按何种机理进行呢?这与卤代烷结构,离去基团亲核试剂和溶剂的性质等诸因素有关,下面分别讨论。 一、烷基结构的影响 1.烷基的结构对S N2 反应的影响 在卤代烷的S N2 反应中,如果中心碳原子上连接的取代烷基(支链)越多,它们对亲核试剂从碳卤键背后进攻中心碳原子的空间位阻就越大,使得发生有效碰撞的概率大为下降;而在过渡态时众多的支链与中心碳原子要保持在同一个平面内,其张力是很大的,这就使形 成过渡态需要有非常高的活化能,这些都将导致卤代烷进行S N2 反应的活性下降,反应速率减小。例如,I-与下面各溴代烷的丙酮溶液中于25℃发生S N2 反应时的相对反应速率为: 如果在卤代烷的β- 碳原子上连有支链烷基时,对S N2 反应的速率也有明显的影响,即卤代烷中心碳( α- 碳)原子上连接的烷基体积越大,其空间位阻越大,不利于亲核试剂的攻 击。例如,在C 2H5 OH 溶剂中C2H5ONa 与下面各溴代烷于55℃发生S N2 反应的相对反应速率为: 反应物CH3CH2B r CH3CH2CH2B r (CH3)2CHCH2B r (CH3)3CCH2B r 相对速 率 100 28 3.0 4.2×10-4 综上所述,卤代烷进行S N2 反应时,在其它条件相同时,不同结构卤代烷的反应活性次序为:
芳香族亲电取代反应的研究 摘要:总结了芳香族亲电取代反应的过程,并对其反应机理进行深入的探讨,对典型的 芳香族亲电取代反应进行了归纳,同时讨论了芳香族化合物的结构和性能的关系。 关键词:亲电取代;反应机理 1芳香族亲电取代反应总论 苯的芳香性结构决定了苯的稳定性。苯环上的:电子为芳香取代反应供给了电子,很多亲电试剂能与之反应,其范围远比烯、炔加成反应广泛。可以用一个统一的反应机理来解释这些取代反应,虽然反应的动力学、自由能—反应曲线对每一种亲电试剂来说可以是不一样的,但若干中间步骤基本上是相似的,这就允许用一个亲电试剂E+做为总的代表反应的一个组成部分,芳环的二体系也参加到反应中去,可以中间体或过渡态的形式出现,有时是一种Π络合物,但它很快转变为。δ络合物,后者以环上的某一个碳与亲电试剂结合,这个碳也就是取代反应发生的位置。δ和Π二络合物的形成和解体是可逆的,从络合物上退减后才得到稳定的芳香体系,究竟退减是H还是E,要看哪一个易于被排除,对大多数取代反应来说,退减一个质子容易的话,这一取代反应是不可逆的,但在某些条件下,如果亲电取代基团是可以被H所排除的,这个反应就是可逆的。δ络合物是反应活性和定位的关键。硝化反应作为不可逆亲电取代反应的代表,已经证明过亲电试剂是硝基正离子NO2十,从硝酸离解出NO2十是决定性的步骤。动力学研究发现有两种速度规律,对于相对不活泼的芳香底物,速度是二级的,硝化试剂一级,芳香化合物也是一级,这相当于亲电试剂和芳香底物的相互结合成为速度最慢步骤.对比较活泼的芳香底物,这一步快于硝基正离子的生成速度,于是速度式中没有芳香化合物浓度一项,在这种情况下,各类芳香性化合物的硝化反应速度相等,都取决于硝酸浓度一项。通常在芳香取代反应中亲电试剂的活性是很大的,所得中间体也很活泼,生成亲电试剂的过程可能是决定反应速度的一步,但也不一定具有决定性的作用。 1.1、反应机理 1.芳环亲电取代反应的一般机理 1.2下列为常见的亲电试剂及其发生的形式:
亲核取代反应及其影响因素 摘要:饱和碳原子上的亲核取代反应主要有两种:单分子亲核取代反应(S N1)与双分子亲核取代反应(S N2)。大多数反应介于这两种极端情况之间。人们提出离子对机理与邻近基团参与的理论来解释反应情况与构型变化的问题。亲核取代反应的反应速度,与烃基的数量、离去集团的大小、亲核试剂的活性以及溶剂的极性等有关。一般来说,烃基数量少,离去基团大,亲核试剂亲核性强,溶剂的极性弱,对S N2反应有利;烃基数量多,离去基团大,溶剂的极性强,对S N1反应有利。另外,亲核取代反应和对应的消除反应(单分子消除反应E1、双分子消除反应E2与S N1、S N2)互为竞争性反应。强碱和较高的温度有利于消除,弱碱和强亲核试剂有利于取代;有利于碳正离子生成的条件,有利于按单分子机理进行;不利于底物异裂的条件,有利于双分子反应。 正文: 一、烷基结构的影响 1.烷基的结构对S N2 反应的影响 反应中,如果中心碳原子上连接的取代烷基(支链)越多,它们对亲在卤代烷的S N2 核试剂从碳卤键背后进攻中心碳原子的空间位阻就越大,使得发生有效碰撞的概率大为下降;而在过渡态时众多的支链与中心碳原子要保持在同一个平面内,其张力是很大的,这就使形 反应的活性下降,反应速成过渡态需要有非常高的活化能,这些都将导致卤代烷进行S N2 率减小。例如,I-与下面各溴代烷的丙酮溶液中于25℃发生S N2 反应时的相对反应速率为: 反应的速率也有明显的影响,即卤如果在卤代烷的β- 碳原子上连有支链烷基时,对S N2 代烷中心碳( α- 碳)原子上连接的烷基体积越大,其空间位阻越大,不利于亲核试剂的攻击。 反应时,在其它条件相同时,不同结构卤代烷的反应活性综上所述,卤代烷进行S N2 次序为:
亲和取代反应总结
亲核取代反应总结 1、反应定义: 亲核取代反应(Nucleophilic Substitution Reaction)是指有机分子中与碳相连的某原子或基团被作为亲核试剂的某原子或基团取代的反应。在反应过程中,取代基团提供形成新键的一对电子,而被取代的基团则带着旧键的一对电子离去。 2、反应意义: 这类反应是有机化学中非常重要的一类反应,不论在理论研究中还是在有机合成实际中都是极其有用的一类反应。 3、反应分类: 亲核取代反应的主要类型为脂肪族饱和碳上的亲核取代反应,即饱和卤代烃与亲核试剂的取代反应,较特殊结构的有苄基卤代物、烯丙基卤代物亲核反应。其他类型还包括与酰氯、磺酸酯、磺酰卤、卤代苯等的取代反应。 从电荷类型来分,亲核取代反应只能有四种类型: (1)中性底物和负离子亲核试剂反应 (2)中性底物和中性亲核试剂反应 (3)正离子底物和负离子亲核试剂反 (4) 正离子底物和中性亲核试剂反应
亲核试剂包括有机和无机两类分子或离子: 无机类亲核试剂:OH-、CN-、X-、H2O、NH3等 有机类亲核试剂:ROH、RO-、PhO-、RS-、RMgX、RCOO-等4、反应机理类型分类: (1)双分子亲核取代反应(S N2) 有两种分子参与了决定反应速率关键步骤的亲核取代反应称为双分子亲核取代反应。反应过程中,亲核试剂从反应物离去基团的背面向与它连接的碳原子进攻,先与碳原子形成比较弱的键,同时离去基团与碳原子的键有一定程度的减弱,两者与碳原子成一条直线,碳原子上另外三个键逐渐由伞形转变成平面,这需要消耗能量,即活化能,当反应进行和达到能量最高状态即过渡态后,亲核试剂与碳原子之间的键开始形成,碳原子与离去基团之间的键断裂,碳原子上三个键由平面向另一边偏转,整个过程犹如大风将雨伞由里向外反转一样,这时就要释放能量,形成产物,S N2反应机理一般式表示为: Nu-+R X [Nuδ-···R···Xδ- ] NuR+X- 例如,溴甲烷与OH-的水解反应:
第11章苯和芳香烃芳香亲电取代反应 一、选择题 1.下面的化合物进行硝化反应的速度顺序是()。[华中科技大学2000研] A.(3)>(4)>(2)>(1) B.(3)>(2)>(4)>(1) C.(4)>(2)>(3)>(1) D.(4)>(3)>(1)>(2) 【答案】A 【解析】给电子基的给电子能力越强,苯环上的硝化反应越快;吸电子基的吸电子能力越强,硝化反应越慢。 2.下面化合物的正确名称是()。[华中科技大学2000研] A.对甲基苯磺酰胺 B.N-甲基对甲苯磺酰胺 C.对甲苯甲基苯磺酰胺 D.甲氨基对甲苯磺酰胺 【答案】B 【解析】取代基中含磺酰胺基,要以苯磺酰胺作为主体命名。
3.苯甲醚在邻位进行硝化反应时,其中间体的极限结构对共振杂化体贡献最大的是()。[天津大学2000;大连理工大学2004研] 【答案】C 【解析】(C)中的正电荷位于与甲氧基相连的碳原子上,甲氧基的给电子效应使正电荷分散,因此该极限结构比其他三种极限结构相对稳定,对共振杂化体的贡献最大。 4.反应的主要产物是()。[武汉大学2001研] (D)(A),(B)等量(E)(A),(C)等量 【答案】B 【解析】氯原子为邻、对位定位基,因生成(A)时的空间位阻较大,故主要产物为(B)。 5.下列化合物,芳环上起亲核取代反应速率最快的是()。[南京大学2003研]
【答案】C 【解析】与氯原子相连的碳原子带的正电荷越多,则亲核取代反应速率越快。硝基为吸电子基,使苯环上邻、对位电子云密度降低,正电荷增多,故(C)亲核反应速率最快。 6.下列化合物有芳香性的是()。[华中科技大学2000研] 【答案】B,C 【解析】(B)项分子中有6个π电子,符合休克尔规则;(C)项中的七元环带一个单位正电荷,五元环带一个单位负电荷,这样七元环和五元环的π电子数都为6个,且在同一平面内,都符合休克尔规则。 7.下列化合物中有芳香性的是()。[中国科学院-中国科学技术大学2001研] 【答案】A 【解析】(A)中有10个π电子数,符合休克尔规则。 8.薁的亲核取代反应容易发生在哪些位置上?()[上海大学2004研]
亲核取代反应及其影响因素 航03班 林三春 2010011556 摘要: 本文分为四部分。第一部分论述了亲核取代反应的组成部分:亲核试剂、离去基团、反应底物,特地列出了常见的亲核试剂、常见的离去基团。第二部分论述了亲核取代反应机理,主要论述了四种:SN1、SN2、离子对机理和邻近基团参与机理,其中还包括各种机理的实验现象验证,以及对反应产物的影响,如对构型的影响。第三部分论述了亲核反应的影响因素,主要有烃基、离去基团、溶剂和亲核试剂四种,详细地说明了这四种因素如何影响反应。给出了判断离去基团的好坏,以及比较亲核试剂的亲核性的方法。最后一部分论述了亲和取代反应与消除反应的竞争关系,其中包括SN1与E1竞争,SN2与E2竞争。主要以卤代烃为例阐述的。 在论述的同时,还附有适当的图示,以及实验数据,通过比较等手段,使得论述更加有说服力。 全文通过这四个部分,详细、全面地介绍了亲核取代反应。 正文: 亲核取代反应,简称SN 亲核取代反应,通常发生在带有正电或部分正电荷的碳上,碳原子被带有负电或部分负电的亲核试剂(Nu:-)进攻而取代。 一、亲核取代反应的重要组成成分: 亲核取代反应中涉及到的三个重要组成成分为:亲核试剂、离去基团、反应底物。 称为反应底物。进攻反应底物的试剂CH30Na (或 CH3O —)是带着电子对与碳原子结合成键的,它本身具有亲核性,称为亲核试剂,一般用Nu 表示。这类反应之所以称为亲核取代也正是因为它是由亲核试剂进攻反应底物而引起的取代反应。反应底物上的溴原子带着电子对从碳原子上离去,所以Br-;称为离去基团,一般用L 表示。该取代反应是在与溴相连的那个碳原子上进行的,常称该碳原子为中心原子,或反应中心。 .一般的亲核取代反应可以用如下的通式表示: 。其中R —L 为反应底物,L —为离去基团,Nu —为亲核试剂,弯箭头表示电子转移的方向。 1、亲核试剂: 亲核性是指:带负电荷或孤对电子的试剂即亲核试剂对亲电子原子的进攻的能力。 具有亲核性的物质叫做亲核试剂。凡是带有未共享电子对的物质(如Lewis 碱)都具有一定的亲核性,它们都可能作为亲核试剂。亲核试剂可以是中性分子,也可以是带负电的阴离子。下表列出的是亲核取代反应中常见的一些亲核试剂:
第七章芳环上的亲电和亲核取代反应7.1芳环的亲电取代反应 7.1.1芳环上的亲电取代历程 1、亲电试剂的产生 亲电试剂 2、π-络合物的形成 芳环上电子云密度R 3、σ-络合物的形成 硝基所在碳为sp3杂化 4、消去-H+ σ-络合物的证据 已分离出C+ 通过红外和核磁等可鉴定中间体的结构。
7.1.2苯环上亲电取代反应的定位规律 从反应速度和取代基进入的位置进行考虑 1、第一类定位基(邻,对位定位基) 2、第二类定位基(间位定位基) 均为致钝基 7.1.3定位规律在有机合成中的应用 7.1.4典型的芳香亲电取代反应 1.硝化反应 硝化试剂有HNO3-H2SO4 真正的硝化试剂为硝酰正离子。混酸体系有强氧化性 如用混酸将得氧化产物 同时还有部分氧化产物HNO3/CCl4低温时的硝化速度较快 温和的硝化试剂HNO2/C(NO2)4
可避免间位硝化与氧化 2.磺化反应 亲电试剂为SO3或(共轭酸) 特点:(1)可逆反应,可用于芳环的定向取代(占位)。(2)置换反应,合成一些难于合成的物质。 发生间接硝化 3.卤化反应 (1)卤素作卤化剂
(2)N-卤代酰胺或N-卤代磺酰胺作卤化剂 等 其卤化性能较差,只与活泼芳烃反应,可避免氧化反应发生(芳胺和酚)。 而在非极性CCl4等溶剂中是自由基引发剂 自由基取代反应。 1. Fridel-Crafts反应 (1) 烃基化 亲电试剂产生 催化剂活性 AlCl3>FeCl3>SbCl5>SnCl4>BF3>TiCl4>ZnCl2 特点 A. 易发生重排反应
亲电试剂发生重排 B. 易发生多烷基化 C. 可逆反应 动力学条件下,遵守定位规律,热力学控制条件下得稳定的间位产物。 D. 钝化的芳烃不发生烷基化 E.-OH,-NH2和-OR等与路易斯酸配位,使催化剂难于发生烷基化,可改用烯作烷基化剂,以酚铝或苯胺铝作催化剂 (2)酰基化反应 1 用酰氯时,ACl3的量要大于1mol,用酸酐时ACl3要大于2mol。 2 酚的酰化是Fries重排 3 不会发生重排 5.重氮盐的偶联反应
芳香类化合物是指从植物胶里取得的具有芳香气味的物质。现在人们把具有下列特殊稳定的不饱和环状化合物称为方向化合物: (1)从结构上看①芳香化合物一般都具有平面或接近平面的环状结构②键长趋于平均化③具有较高的C/H 比 (2)从性质上看①芳香化合物的芳环一般都难以氧化、加成②而易于发生亲电取代反应③具有特殊的光学性质,如芳环环外氢的化学位移处于NMR 低场,而环内氢处于高场等。 上述这些特点被称为芳香性。 1. 芳香烃的结构 1.1苯的结构和表达 1.1.1苯的结构 第十一章 苯和芳香烃 芳香亲电取代反应 120o 108pm 140pm
1.1.2苯的芳香性 环己烯的氢化热ΔH=-120kJ/mol,1,3-环己二烯的氢化热ΔH=-232kJ/mol(由于其共轭双键增加了其稳定性)。而苯的氢化热ΔH=-208kJ/mol。1,3-环己二烯失去两个氢变成苯时,不但不吸热,反而放出少量的热量。这说明:苯比相应的环己三烯类要稳定得多,从1,3-环己二烯变成苯时,分子结构已发生了根本的变化,并导致了一个稳定体系的形成。 苯难于氧化和加成,而易于发生亲电取代反应,与普通烯烃的性质有明显的区别。 1.2.3苯的表达 自1825年英国物理学家和化学家Farady M(法拉第)首先从照明气中分离出苯后,人们一直在探索苯结构的表达式。科学家们提出了各种有关苯结构式的假设;其中比较有代表性的苯的结构式有: 凯库勒式双环结构式棱形结构式向心结构式对位键结构式余键结构式 (凯库勒(杜瓦(拉敦保格(阿姆斯特朗、拜耳(克劳斯(悌勒 1865年提出)1866-1867年提出)1869年提出)1887-1888年提出)1888年提出)1899年提出)用内部带有一个圆圈的正六边形表示苯,圆圈强调了π电子的离域作用和电子云的均匀分布,它很好地说明了 碳碳键键长的均等性和苯环的完全对称性,但是这种方式用来表示其他方向体系时就不合适了,例如表示萘时很容 易造成误解,因为萘不是完全对称的分子,萘分子的碳碳键长也不是完全均等的。另外,圆圈没有说明π电子的数 目,萘分子的10个π电子用两个圆圈表示易误解成每个环有6个π电子而造成混淆。
亲核取代反应 一.亲核取代反应机理。 亲核取代反应是指有机分子中的与碳相连的原子或原子团被作为亲核试剂的某原子或原子团取代的反应。反应分为SN1型(单分子取代反应),与SN2型双分子取代反应。 1.SN1型(单分子取代反应) 第一步是碳原子上正电荷增加,离去基团负点性增加,经过过渡态(1)并最终解离,生成活性中间体碳正离子与离去基团负离子。由于这一步反应的活化能较高,速率较慢,所以这一步是反应的决速步。 第二步是活性中间体的碳正离子与亲和试剂作用,生成反应产物。这一步仅需少量能量,速率很快。 反应特点: (1)SN1反应的决速步是中心碳原子与离去基团之间化学键的异裂。反应速率只取决 于一种分子的浓度,因此,它在动力学上是一级反应。 (2)一般是一个两步反应。第一步生成的碳正离子采取SP2杂化,是平面构型。故 若反应物的中心碳原子是手性碳,反应产物一般是一对等量的对映异构体的混 合物——外消旋体。 (3)反应中间体生成的碳正离子导致反应有重排的趋势。 2.SN2型(双分子取代反应) 反应中,离去基团离开中心碳原子的同时,亲核试剂与中心碳原子发生部分键合,无中间体生成。 有机反应中,将两种分子参与决速步的亲核取代反应陈伟双分子亲核取代反应。 反应特点: (1)SN2反应是一步反应,只有一个过渡态。 (2)在SN2反应中,亲核试剂进攻中心碳原子是总是从离去基团溴原子的背面沿着 碳原子和离去基团连接的中心线方向进攻。这个过程会使得碳原子与三个未参 与反映的键发生翻转,这种翻转称为瓦尔登翻转,又称构型翻转。 二.影响亲核取代反应的因素 1.烃基结构的影响。 对SN1反应,主要考虑碳正离子的稳定性。对SN2反应,主要取决于过渡态形 成的难易,也就是空间效应的影响。 2.离去基团的影响。
亲电取代反应 亲电取代反应是亲电试剂进攻化合物负电部分,取代其它基团的化学反应。一般发生于芳香族化合物,是一种向芳香环系引入官能团的重要方法,是芳香族化合物的特性之一。被取代的基团通常是氢原子,但其他基团被取代的情形也是存在的。一般来说,亲电取代特指芳香亲电取代。另一种比较少见的亲电取代反应是脂肪族的亲电取代。 中文名 亲电取代反应 外文名 Electrophilic Substitution 属性 亲电取代 性质 反应 主要反应 硝化反应,卤化反应磺化反应等。 目录 .1原理
.2主要反应 .?硝化反应 .?卤化反应 .?磺化反应 .3定位规则 原理 亲电取代反应主要发生在芳香体系或富电子的不饱和碳上,就本质而言均是较强亲电基团对负电子体系进攻,取代较弱亲电基团。但对于芳香体系和脂肪体系,由于具体环境不同,其反应历程亦有所不同,现分述如下。 亲电芳香取代反应(electrophilic aromatic substitution)是芳香体系最重要的有机反应之一,常用于向芳香环系引入官能团,因此研究时间较长,在机理方面已基本达成一致。 主要反应 对于亲电取代反应,其最为主要的反应类型均在芳香体系中产生,所以这里仅仅对芳香亲电取代进行一定的举例介绍。 硝化反应 硝化反应苯环体系一个重要的反应,其常用于向体系引入硝基或利用硝基引入氨基等其他各种官能团,有很强的泛用性,定位选择性较好,使用最多。由于硝基有较强氧化性,而有机体系本身又具有一定的还原性,硝基含量较多的体系就很容易成为良好的炸药材料,其中著名的TNT、苦味酸等就是通过硝化反应制备的。 Friedel(傅瑞德尔)-Crafts(克拉夫茨)反应