
4期吴永升等:植物数量性状全基因组选择研究进展1511 全基因组选择的概念和原理 全基因组选择(Genome-wideselection,GWS),又称基因组选择(Genomicselection,GS),由Meu—wissen于2001年首先提出∞J。主要是通过全基因组中大量的分子标记和参照群体(trainingpopula—tion)的表型数据建立BLUP模型估计出每一标记的育种值,然后仅利用同样的分子标记估计出后代个体育种值并进行选择[7】。 全基因组选择理论主要利用连锁不平衡信息,即假设标记与其相邻的QTL处于连锁不平衡状态,因而由相同标记估计的不同群体的染色体片段效应是相同的,这就要求标记密度足够高以使所有的QTL与标记处于连锁不平衡(LD)状态哺J。而目前随着拟南芥、水稻、玉米等植物基因组序列图谱及SNP图谱的完成或即将完成,提供了大量的SNP标记用于基因组研究。而随着SNP芯片等大规模高通量SNP检测技术的发展和成本的降低,使得全基因组选择应用成为可能。 2全基因组选择的基本方法及案例说明 2.1全基因组选择的基本方法 全基因组选择在实施过程中应该包括以下几个基本步骤:在需要实行选择的参照群体中获取参照群体的基因型数据和表现型数据;然后,通过BLUP程序估计出每个标记位点的标记效应值,从而获得育种值;最后,在接下来每一轮的选择中,不再需要表型数据,根据每一轮次群体基因型信息估计育种值,直接选择群体的优良单株【9j。 全基因组选择的核心过程就是用从参照群体中每一个体的表现型数据和基因型数据建立的数学模型来估算接下来的育种群体中仅有基因型数据的个体的GEBV值。由既有表现型数据又有基因型数据的每一个体组成的群体被成为参照群体。参照群体用来估计数学模型的参数,这个参数接着用来计算仅有基因型数据的育种个体GEBV值,然后根据计算的GEBV值对育种群体进行选择并提升到下一轮次的选择中。因此,通过模型来预测个体的育种值,可以不进行表型鉴定就直接对育种群体的个体进行选择(Meuvissen,2001)。为了使估算的GEBV值尽可能地准确,参照群体必须具有代表性,尽可能地代表接下来在育种过程中用全基因组选择方法来进行选择的分离群体。 2.2全基因组选择方法案例 如图l所示,在这个例子中,笔者的目标是把外来种质中的优良性状基因(包括产量、矮杆、抗逆等)导入本地优良的自交系,从而实现种质的改良 图1在玉米中利用全基因组选择方法导入外源种质 Fig.1Genomewideselectionto introgr%exotictraitsintoadaptedmaize
陆地棉基因组测序揭示四倍体棉进化与纤维发育机制Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement 研究对象:陆地棉遗传标准系TM-1 期刊:Nature Biotechnology 影响因子:41.514 合作单位:南京农业大学 发表时间:2015年4月 摘 要 Upland cotton is a model for polyploid crop domestication and transgenic improvement. Here we sequenced the allotetraploid Gossypium hirsutum L. acc. TM-1 genome by integrating whole-genome shotgun reads, bacterial artificial chromosome (BAC)-end sequences and genotype-by-sequencing genetic maps. We assembled and annotated 32,032 A-subgenome genes and 34,402 D-subgenome genes. Structural rearrangements, gene loss, disrupted genes and sequence divergence were more common in the A subgenome than in the D subgenome, suggesting asymmetric evolution. However, no genome-wide expression dominance was found between the subgenomes. Genomic signatures of selection and domestication are associated with positively selected genes (PSGs) for fiber improvement in the A subgenome and for stress tolerance in the D subgenome. This draft genome sequence provides a resource for engineering superior cotton lines.关键词 陆地棉;de novo;四倍体 研究背景 陆地棉(Gossypium hirsutum L.)隶属锦葵目(Malvales),锦葵科(Malvaceae),棉属(Gossypium),因最早在美洲大陆种植而得名,是世界上最重要的棉花栽培品种,占全球棉花种植面积的90%以上。尽管陆地棉在棉花产业中占据核心地位,但由于其为异源四倍体,相关的全基因组测序工作一直难以开展。来自南京农业大学、北京诺禾致源、美国德克斯大学的国际团队,利用最新测序技术,成功构建了高质量的陆地棉全基因组图谱,为进一步改良棉花的农艺性状提供了基础,同时也为多倍体植物的形成和演化机制提供了新的启示。
第9卷 第1期2007年3月 衡水学院学报 J o u r n a l o f H e n g s h u i U n i v e r s i t y V o l.9,N o.1 Ma r.2007植物功能基因组学及其研究技术 崔兴国 (衡水学院 生命科学系,河北 衡水053000) 摘 要:植物基因组的研究已经由以全基因组测序为目标的结构基因组学转向以基因功能鉴定为目标的功能基因组学研究.植物功能基因组学研究是利用结构基因组学积累的数据,从中得到有价值的信息,阐述D N A序列的功能,从而对所有基因如何行使其职能并控制各种生命现象的问题作出回答.近年来植物功能基因组学的研究技术主要包括表达序列标签、基因表达的系列分析、D N A微阵列和反向遗传学等.对植物功能基因组学的研究将有利于我们对基因功能的理解和对植物形状的定性改造和利用. 关键词:植物;功能基因组学;研究技术 中图分类号:Q3-3 文献标识码:A 文章编号:1673-2065(2007)01-0023-04 基因是细胞的遗传物质,决定细胞的生物学形状,细胞的生物学功能最终是由大量的基因表达完成的.随着人类基因组“工作框架图”的完成,生命科学研究的重点已经从结构基因组学转移到了功能基因组学的研究,特别是模式植物拟南芥(A r a b i d o p-s i s t h a l i a n a)和水稻(O r y z a s a t i v a)基因组测序的完成,公共数据库中已经积累了大量基因序列信息,获得了许多与植物发育相关的功能基因,在此基础上应用实验分析方法并结合统计和计算机分析来研究基因的表达、调控与功能,并相应诞生和发展了一批新的研究技术,为功能基因组学的研究提供了必要而有效的技术支撑.功能基因组学研究的最终目标是解析所有基因的功能,即从基因水平上大规模批量鉴定基因的功能,进而全面研究控制植物生长发育及响应环境变化的遗传机制,在基因组序列与细胞学行为之间起到桥梁作用,共同承担起从整体水平上解析生命现象的重任. 1 植物功能基因组学研究 植物的生长和发育是一个有机体或有机体的一部分形态建成和功能按一定次序而进行的一系列生化代谢反应的总合,反应在分子水平上,它要求相应的遗传代谢途径必须按照特定的时空次序严格进行以保证正常发育.植物功能基因组研究就是要利用植物全基因组序列的信息,通过发展和应用系统基因组水平的实验方法来研究和鉴别基因组序列的作用;研究基因组的结构、组织与植物功能在细胞、有机体和进化上的关系以及基因与基因间的调控关系;从表达时间、表达部位和表达水平3个方面对目的基因在植物中的精细调控进行系统研究.当前植物功能基因组学研究主要集中于一年生的拟南芥与水稻两个物种上,这主要是由于它们的遗传背景清楚,基因组较小,基因结构简单而且易于进行分子生物学操作.拟南芥研究组“2010计划”的宏伟目标是充分利用拟南芥基因组计划获得的序列信息并结合功能基因组研究技术来获知其25000个基因的全部功能,例如开花的诱导过程是植物生活周期中最奇妙的过程,目前从拟南芥中鉴定了提早开花和延迟开花的多种突变体,显示植物开花受多个遗传基因的控制,如延迟开花的两个突变体是由等位基因 C O(C O N S T A N S)和L D(C O L D L U M I N I D E P E N- D E N S)突变引起,这两个基因均已被克隆,并使其在转基因植物的叶片中进行表达,将C O基因转移到拟南芥中,高效表达C O蛋白的转基因植株即使处于短日照条件下也会开花,这说明C O基因具有激活开花基因的作用.对模式植物功能基因组的研究将有助于整个植物基因组学的研究. 目前的功能基因组研究主要包括以下几个方面:(1)c D N A全长克隆与测序;(2)获得D N A芯片 ①收稿日期:2006-10-12 作者简介:崔兴国(1963-),女,河北冀州市人,衡水学院生命科学系副教授.
水稻、玉米、大豆、甘蓝、白菜、高粱、黄瓜、西瓜、马铃薯、番茄、拟南芥、杨树、麻风树、苹果、桃、葡萄、花生 拟南芥籼稻粳稻葡萄番木瓜高粱黄瓜玉米栽培大豆苹果蓖麻野草莓马铃薯白菜野生番茄番茄梨甜瓜香蕉亚麻大麦普通小麦西瓜甜橙陆地棉梅毛竹桃芝麻杨树麻风树卷柏狗尾草属花生甘蓝 物种基因组大小和开放阅读框文献 Sesamum indicum L. Sesame 芝麻(2n = 26)293.7 Mb, 10,656 orfs 1 Oryza brachyantha短药野生稻261 Mb, 32,038 orfs 2 Chondrus crispus Red seaweed爱尔兰海藻105 Mb, 9,606 orfs 3 Pyropia yezoensis susabi-nori海苔43 Mb, 10,327 orfs 4 Prunus persica Peach 桃226.6 of 265 Mb 27,852 orfs 5 Aegilops tauschii 山羊草(DD)4.23 Gb (97% of the 4.36), 43,150 orfs 6 Triticum urartu 乌拉尔图小麦(AA)4.66 Gb (94.3 % of 4.94 Gb, 34,879 orfs 7 moso bamboo (Phyllostachys heterocycla) 毛竹2.05 Gb (95%) 31,987 orfs 8 Cicer arietinum Chickpea鹰嘴豆~738-Mb,28,269 orfs 9 520 Mb (70% of 740 Mb), 27,571 orfs 10 Prunus mume 梅280 Mb, 31,390 orfs 11 Gossypium hirsutum L.陆地棉2.425 Gb 12 Gossypium hirsutum L. 雷蒙德氏棉761.8?Mb 13 Citrus sinensis甜橙87.3% of ~367 Mb, 29,445 orfs 14 甜橙367 Mb 15 Citrullus lanatus watermelon 西瓜353.5 of ~425 Mb (83.2%) 23,440 orfs 16 Betula nana dwarf birch,矮桦450 Mb 17
川金丝猴全基因组测序解析其植食性机制与进化史 Whole-genome sequencing of the snub-nosed monkey provides insights into folivory and evolutionary history 研究对象:川金丝猴期刊:Nature Genetics 影响因子:29.352 合作单位:中国科学院动物研究所发表时间:2014年11月 摘 要 Colobines are a unique group of Old World monkeys that principally eat leaves and seeds rather than fruits and insects. We report the sequencing at 146× coverage, de novo assembly and analyses of the genome of a male golden snub-nosed monkey (Rhinopithecus roxellana ) and resequencing at 30× coverage of three related species (Rhinopithecus bieti , Rhinopithecus brelichi and Rhinopithecus strykeri ). Comparative analyses showed that Asian colobines have an enhanced ability to derive energy from fatty acids and to degrade xenobiotics. We found evidence for functional evolution in the colobine RNASE1 gene, encoding a key secretory RNase that digests the high concentrations of bacterial RNA derived from symbiotic microflora. Demographic reconstructions indicated that the profile of ancient effective population sizes for R. roxellana more closely resembles that of giant panda rather than its congeners. These findings offer new insights into the dietary adaptations and evolutionary history of colobine primates. 关键词 金丝猴;重测序;植食性;进化 研究背景 金丝猴(Rhinopithecu spp.)隶属于灵长目(Primates)、疣猴亚科(Colobinae)、仰鼻猴属 (Rhinopithecus ) ,目前共有5个种,即川、滇、黔、缅甸和越南金丝猴。通过对金丝猴基因组以及肠道基因组进行全面系统的研究,解析了金丝猴的植食性分子遗传机制,为了解疣猴亚科的系统进化、功能适应性奠定了遗传基础,同时开展了仰鼻猴属的进化历史和遗传多态性分析。
千年基因将应邀参加第十六届全国植物基因组学大会 第十六届全国植物基因组学大会将于2015年8月19日-22日在陕西杨凌召开,千年基因应邀参加此次会议,并将在会场学术交流区设立展台。届时千年基因的技术团队会向大家展示我们最全面的测序平台、一站式的基因组学解决方案以及近年来在植物基因组学领域取得的科研成果,欢迎广大科研人员莅临指导交流! 在测序平台方面,千年基因目前拥有国内最全面的测序平台,能够为科研人员提供一站式解决方案。以PacBio RS II三代平台为例,千年基因自去年提供PacBio RS II测序以来,通过项目经验的积累及严格的质量控制,目前各项数据指标已达国内最高水平。数据产出已稳步升级至1.4Gb/ SMRT cell,读长最长可达42 Kb,reads N50高达18Kb,远超PacBio官方提供的数据标准!在植物基因组de novo测序的研究中,千年基因提供的超长读长测序可更好地跨越基因组高重复序列、转座子区域以及大的拷贝数变异区域和结构变异区,从而实现对高杂合及高重复基因组的完美组装。在植物转录组测序的研究中,千年基因提供的超长读长测序无需拼接即可获得全长转录组序列信息,同时可获得全面的可变剪切、融合基因以及Isoform信息。另外,千年基因提供的HiSeq 4000及HiSeq 2000/2500测序可解决研究人员在植物基因组重测序、转录组测序、小RNA测序等方面的科研需求。 在项目经验方面,千年基因与来自全球的科研人员合作开展了大量植物基因组项目,相关成果已发表于Nature、Nature Genetics、Science等杂志。例如,油棕榈基因组项目在Nature 杂志同时发表两篇文章,辣椒基因组项目的成果发表于Nature Genetics,玉米基因组项目的成果发表于Science。在国外合作方面,千年基因与美国爱荷华州立大学Patrick Schnable教授领导的国际玉米基因组团队合作开展的上万份玉米样本重测序项目也正在进行中;千年基因与国际半干旱热带作物研究所建立长期战略合作关系,正在开展上千份木豆、鹰嘴豆及高粱样本的群体遗传学研究;同时千年基因与华盛顿大学的Evan Eugene Eichler院士及佐治亚大学的Jeffrey Lynn Bennetzen院士也有大量基因组项目合作。在国内合作方面,千年基因与广东省农科院、山东省农科院共同启动的花生基因组项目已全部完成de novo测序及数据挖掘,同时与中国科学院、北京大学、中国农业大学、中国科学技术大学、上海交通大学、
全基因组从头测序(de novo测序) https://www.doczj.com/doc/be15592076.html,/view/351686f19e3143323968936a.html 从头测序即de novo 测序,不需要任何参考序列资料即可对某个物种进行测序,用生物信息学分析方法进行拼接、组装,从而获得该物种的基因组序列图谱。利用全基因组从头测序技术,可以获得动物、植物、细菌、真菌的全基因组序列,从而推进该物种的研究。一个物种基因组序列图谱的完成,意味着这个物种学科和产业的新开端!这也将带动这个物种下游一系列研究的开展。全基因组序列图谱完成后,可以构建该物种的基因组数据库,为该物种的后基因组学研究搭建一个高效的平台;为后续的基因挖掘、功能验证提供DNA序列信息。华大科技利用新一代高通量测序技术,可以高效、低成本地完成所有物种的基因组序列图谱。包括研究内容、案例、技术流程、技术参数等,摘自深圳华大科技网站 https://www.doczj.com/doc/be15592076.html,/service-solutions/ngs/genomics/de-novo-sequencing/ 技术优势: 高通量测序:效率高,成本低;高深度测序:准确率高;全球领先的基因组组装软件:采用华大基因研究院自主研发的SOAPdenovo软件;经验丰富:华大科技已经成功完成上百个物种的全基因组从头测序。 研究内容: 基因组组装■K-mer分析以及基因组大小估计;■基因组杂合模拟(出现杂合时使用); ■初步组装;■GC-Depth分布分析;■测序深 度分析。基因组注释■Repeat注释; ■基因预测;■基因功能注释;■ ncRNA 注释。动植物进化分析■基因家族鉴定(动物TreeFam;植物OrthoMCL);■物种系统发育树构建; ■物种分歧时间估算(需要标定时间信息);■基因组共线性分析; ■全基因组复制分析(动物WGAC;植物WGD)。微生物高级分析 ■基因组圈图;■共线性分析;■基因家族分析; ■CRISPR预测;■基因岛预测(毒力岛); ■前噬菌体预测;■分泌蛋白预测。 熊猫基因组图谱Nature. 2010.463:311-317. 案例描述 大熊猫有21对染色体,基因组大小2.4 Gb,重复序列含量36%,基因2万多个。熊猫基因组图谱是世界上第一个完全采用新一代测序技术完成的基因组图谱,样品取自北京奥运会吉祥物大熊猫“晶晶”。部分研究成果测序分析结果表明,大熊猫不喜欢吃肉主要是因为T1R1基因失活,无法感觉到肉的鲜味。大熊猫基因组仍然具备很高的杂合率,从而推断具有较高的遗传多态性,不会濒于灭绝。研究人员全面掌握了大熊猫的基因资源,对其在分子水平上的保护具有重要意义。 黄瓜基因组图谱黄三文, 李瑞强, 王俊等. Nature Genetics. 2009. 案例描述国际黄瓜基因组计划是由中国农业科学院蔬菜花卉研究所于2007年初发起并组织,并由深圳华大基因研究院承担基因组测序和组装等技术工作。部分研究成果黄瓜基因组是世界上第一个蔬菜作物的基因组图谱。该项目首次将传
生物五界:动物、植物、真菌、原生生物和原核生物;生物三界:真细菌、古细菌、真核生物 具有催化活性的RNA分子称为核酶(ribozyme)核酶催化的生化反应有:自我剪接、催化切断其它RNA、合成多肽键、催化核苷酸的合成 新基因的产生:基因与基因组加倍1)整个基因组加倍; 2)单条或部分染色体加倍;3)单个或成群基因加倍。DNA水平转移:原核生物中的DNA水平转移可通过接合转移,噬菌体转染,外源DNA的摄取等不同途径发生,水平转移的基因大多为非必须基因。动物中由于种间隔离不易进行种间杂交,但其主要来源于真核细胞与原核细胞的内共生。动物种间基因转移主要集中在逆转录病毒及其转座成分。 外显子洗牌与蛋白质创新:产生全新功能蛋白质的方式有二种:功能域加倍,功能域或外显子洗牌 基因冗余:一条染色体上出现一个基因的很多复份(复本)当人们分离到某一新基因时,为了鉴定其生物学功能,常常使其失活,然后观察它们对表型的影响。许多场合,由于第二个重复的功能基因可取代失活的基因而使突变型表型保持正常。这意味着,基因组中有冗余基因存在。看家基因很少重复,它们之间必需保持剂量平衡,因此重复的拷贝很快被淘汰。与个体发育调控相关的基因表达为转录因子,具有多功能域的结构。这类基因重复拷贝变异可使其获得不同的表达控制模式,促使细胞的分化与多样性的产生,并导致复杂形态的建成,具有许多冗余基因。 非编码序列扩张方式:滑序复制、转座因子 模式生物海胆、果蝇、斑马鱼、线虫、蟾蜍、小鼠、酵母、水稻、拟南芥等。模式生物基因组中G+C%含量高, 同时CpG 岛的比例也高。进化程度越高, G+C 含量和CpG 岛的比例就比较低 如果基因之间不存在重叠顺序,也无基因内基因(gene-within-gene),那么ORF阅读出现差错的可能只会发生在非编码区。细菌基因组中缺少内含子,非编码序列仅占11%, 对阅读框的排查干扰较少。细菌基因组的ORF阅读相对比较简单,错误的机率较少。高等真核生物DNA的ORF阅读比较复杂:基因间存在大量非编码序列(人类占70%);绝大多数基因内含有非编码的内含子。高等真核生物多数外显子的长度少于100个密码子 内含子和外显子序列上的差异:内含子的碱基代换很少受自然选择的压力,保留了较多突变。由于碱基突变趋势大多为C-T,故A/T的含量内含子高于外显子。由于终止密码子为TAA\TAG\TGA,如果以内含子作为编码序列,3种读码框有很高比例的终止密码子。 基因注释程序编写的依据:1)信号指令,包括起始密码子,终止密码子,终止信号,剪接受体位和供体位,多聚嘧啶序列,分支点保守序列2)内容指令,密码子偏好,内含子和外显子长短 基因功能的检测:基因失活、基因过表达、RNAi干涉 双链DNA的测序可从一端开始,亦可从两端进行,前者称单向测序,后者称双向测序。 要获得大于50 kb的DNA限制性片段必需采用稀有切点限制酶。 酵母人工染色体(YAC)1)着丝粒在细胞分裂时负责染色体均等分配。2)端粒位于染色体端部的特异DNA序列,保持人工染色体的稳定性3)自主复制起始点( ARS)在细胞中启动染色体的复制 合格的STS要满足2个条件:它应是一段序列已知的片段,可据此设计PCR反应来检测不同的DNA片段中是否存在这一顺序;STS必需在染色体上有独一无二的位置。如果某一STS在基因组中多个位点出现,那么由此得出的作图数据将是含混不清的。 遗传图绘制主要依据由孟德尔描述的遗传学原理,第一条定律为等位基因随机分离,第二条定律为非等位基因自由组合,显隐性规律/不完全显性、共显性、连锁 衡量遗传图谱的水平覆盖程度饱和程度 基因类型:transcribed, translatable gene (蛋白基因 ) ; transcribed but non-translatable gene ( RNA 基因 )Non- transcribed, non-translatablegene ( promoter, operator ) rRNA基因,tRNA基因, scRNA基因, snRNA 基因, snoRNA基因, microRNA基因 基因组(genome):生物所具有的携带遗传信息的遗传物质总和。 基因组学(genomic):用于概括涉及基因作图、测序和整个基因功能分析的遗传学分支。 染色体组(chromosome set):不同真核生物核基因组均由一定数目的染色体组成,单倍体细胞所含有的全套染色体。 比较基因组学(comparative genomics):比较基因组学是基因组学与生物信息学的一个重要分支。通过模式生物基因组与人类基因组之间的比较与鉴别,为分离重要的候选基因,预测新的基因功能,研究生物进化提供依据。(目标)
动物基因组DNA的提取 [实验原理] 在EDTA和SDS等去污剂存在下,用蛋白酶K消化细胞,随后用酚抽提,可以得到哺乳动构基因组DNA,用此方法得到的DNA长度为100-150 kb,适用于L嘴菌体构建基因组文库和Southern分析。 通过本实验了解并掌握提取基因组DNA的原理和步骤,以及相对分子质量较大的DNA 的琼脂糖凝胶电泳技术。 [仪器、材料与试剂] (一)仪器 1.台式离心机 2.玻璃匀浆器 3.高压灭菌锅 4.恒温水浴 (二)材料 1.1.5mL微量离心管 2.微量取样器和吸头 3.无菌过滤器(一次性) 4.10 mL注射器 5.鼠肝 6.三羟甲基氨基甲烷(Tris) 7.十二烷基硫酸钠(SDS) 8.乙二胺四乙酸(EDTA)
9.蛋白酶K 10.RNA酶 11.DNA相对分子质量标准物,DNA/EcoRI+HindⅢ相对分子质量标准物 (三)试剂 1、1.5 mol/L NaCl 2、0.5 mol/L Tris·HCI pH8.0 3.0.5 mol/L EDTA pH8.0 4.3 mol/L NaAc pH5.2 以上均高压灭菌。 5.蛋白酶K 10mg/mL配好后用一次性过滤器过滤,-20 保存(教师配制) 6.组织匀浆液100mmol/LNaCI,10mmol/LTris·HCl(pH 8.0),0.25mmol/LEDTA(pH8.0) 7.酶解液200mmol/LNaCI,20mmol/L Tris·HCI(pH 8.O),50mmol/LEDTA(PH 8.0),200~g/mL蛋白酶K,1%SDS 8.无DNA酵的RNA酶:将胰RNA酶溶解于10mmol/L Tris.HCI(pH7.5)、15 mmol /L NaCl溶液中,浓度l0mg/mL,于100℃水浴处理15min,以降解DNA酶,缓慢冷却到室温,-20℃保存 9.TE缓冲液:10mmol/LTris·HCl(pH8.0),25 mmol/LEDTA(pH8.0) 10.平衡酚(pH8.0):氧仿:异戊醇=25:24:1<体积比) 11.氧仿:异戊醇=24:l(体积比) 12.5xTBE 5.4gTris,2.75g硼酸2mL 0.5mol/L EDTA(pH8.0),加水到100mL;13.6x上样缓冲液o.25%溴酚蓝,40%(W/V)蔗糖水溶液
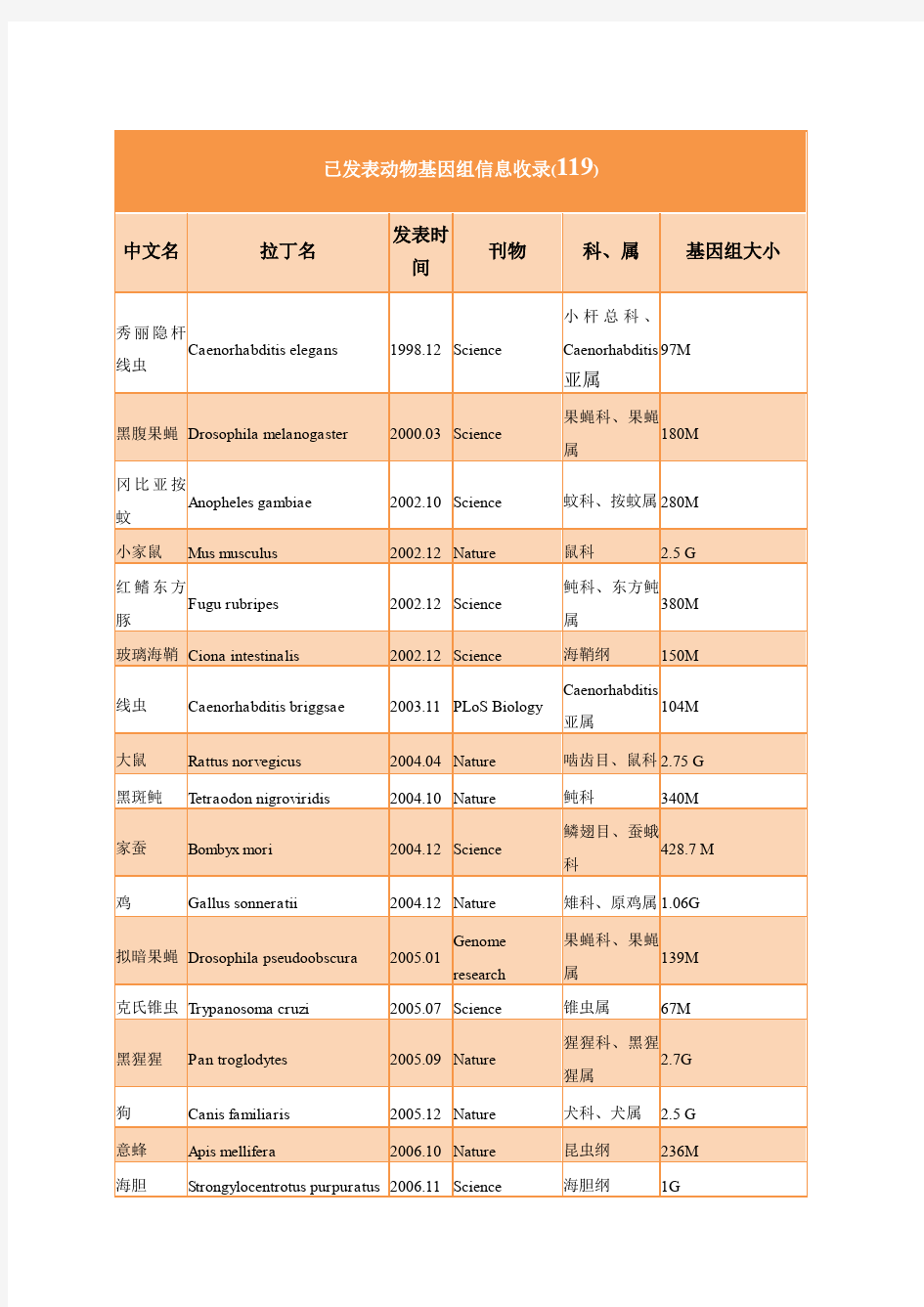
已完成植物基因组测序情况(更新至2014年11月) 中文名拉丁名发表时间刊物科、属基因组大小拟南芥Arabidopsis thaliana 2000.12 Nature 十字花科、鼠耳芥属125M 水稻Oryza sativa. ssp. indica 2002.04 Science 禾本科、稻属466M 水稻Oryza sativa. ssp. japonica 2002.04 Science 禾本科、稻属466M 杨树Populus trichocarpa 2006.09 Science 杨柳科、杨属480M 葡萄Vitis vinifera 2007.09 Nature 葡萄科、葡萄属490M 衣藻Chlamydomonas reinhardtii 2007.01 Science 衣藻科、衣藻属130 M 小立碗藓Physcomitrella pattens 2008.01 Science 葫芦藓科、小立碗藓属480M 番木瓜Carica papaya 2008.04 Nature 番木瓜科、番木瓜属370M 百脉根Lotus japonicus 2008.05 DNA Res. 豆科472 Mb 三角褐指藻Phaeodactylum tricornutum 2008.11 Nature 褐指藻属27.4M 高粱Sorghum bicolor 2009.01 Nature 禾本科、高粱属730M 玉米Zea mays ssp. mays 2009.11 Science 禾本科、玉米属2300M 黄瓜Cucumis sativus 2009.11 Nature Genetics 葫芦科、黄瓜属350M 大豆Glycine max 2010.01 Nature 豆科、大豆属1100M 二穗短柄草Brachypodium distachyon 2010.02 Nature 禾本科、短柄草属260M 褐藻Ectocarpus 2010.06 Nature 水云属196M 团藻Volvox carteri 2010.07 Science 团藻属138M 蓖麻Ricinus communis 2010.08 Nature Biotechnology 大戟科、蓖麻属350M 小球藻Chlorella variabilis 2010.09 Plant Cell 小球藻科46M 苹果Malus × domestica 2010.09 Nature Genetics 蔷薇科、苹果属742M 森林草莓Fragaria vesca 2010.12 Nature Genetics 蔷薇科、草莓属240M 可可树Theobroma cacao 2010.12 Nature Genetics 梧桐科、可可属430-Mb 野生大豆Glycine soja 2010.12 PNAS 豆科、大豆属915.4 Mb 褐潮藻类Aureococcus anophagefferens 2011.02 PNAS 57M 麻风树Jatropha curcas 2010.12 DNA Res. 大戟科、麻风树属410M 卷柏Selaginella moellendorffii 2011.05 Science 卷柏属212M 枣椰树Phoenix dactylifera 2011.05 Nature biotechnology 棕榈科685M 琴叶拟南 芥 Arabidopsis lyrata 2011.05 Nature Genetics 十字花科、鼠耳芥属206.7 Mb 马铃薯Solanum tuberosum 2011.07 Nature 茄目、茄科、茄属844M 条叶蓝芥Thellugiella parvula 2011.08 Nature Genetics 盐芥属140M
植物功能基因组学概述 XXX* (XXXXX) 摘要:植物功能基因组学是从整体水平研究基因的功能及表达规律的科学。对植物功能基因组学的研究将助于我们对基因功能的理解和对植物性状的定性改造和利用。本文简要介绍了植物功能基因组学的概念、研究内容和研究方法。 关键词:植物;功能基因组学;ESTs;SAGE Summarize of Plant Functional Genomics XXX (XXXXX) Abstract:Plant functional genomics studies provide a novel approach to the identification of genome-wide gene expression. It is currently being widely focused on the gene expression by transcript profiling and takes us rapidly forward in our understanding of plant biological traits. In this review, comprehensive of concepts, research contents and methodologies regarding plant functional genomics and transcript profiling are described. Key words: Plant; functional genomics; ESTs; SAGE 1 植物功能基因组学 基因组学(Genomics)是20世纪最后10年研究最活跃的领域之一。基因组学是指对所有基因的结构和功能进行分析的一门学科, 1986年由美国科学家Thomas Roderick提出, 兴起于20世纪90年代[1]。基因组学研究分为结构基因组学( structural genomics) 和功能基因组学( functional genomics)。结构基因组学代表基因组分析的早期阶段, 以建立生物体高分辨率遗传、物理和转录图谱为主, 以研究基因序列为目标。功能基因组学(Functional genomics)的研究又被称为后基因组学(Post genomics)研究,它是利用结构基因组学提供的信息和产物,通过在基因组或系统水平上全面分析基因的功能,使得生物学研究从对单一基因或蛋白质的研究转向对多个基因或蛋白质同时进行系统研究。 植物功能基因组学是植物后基因时代研究的核心内容,它强调发展和应用整体的(基因 组水平或系统水平)实验方法分析基因组序列信息、阐明基因功能,其特点是采用高通量的实验方法结合大规模的数据统计计算方法进行研究。基本策略是从研究单一基因或蛋白质上升到从系统角度研究所有基因或蛋白质。在植物功能基因组学的研究中,拟南芥和水稻是两种最常用的模式植物。目前, 功能基因组学在水稻、拟南芥等模式植物中取得了较快进展, 主要原因在于这两种植物已完成全基因组测序工作[2], 获得了结构基因组数据, 且遗传背景清楚, 易于开展分子生物学研究, 已率先步入后基因组时代。 2 植物功能基因组学研究内容 2、1基因组多样性研究[1] *联系人Tel:XXXXX;E-mail:XXXXX
动植物基因组de novo常见问题 基础知识 1、什么是基因组de novo测序 答:对某一物种进行高通量测序,利用高性能计算平台和生物信息学方法,在不依赖于参考基因组的情况下进行组装,从而绘制该物种的全基因组序列图谱。 2、普通基因组的定义 答:单倍体,纯合二倍体或者杂合度<%,且重复序列含量<50%,GC 含量为35%到65%之间的二倍体。 3、复杂基因组的定义 答:杂合率>%,重复序列含量>50%,GC含量处于异常的范围(GC 含量<35%或者GC含量>65%=的二倍体,多倍体。 诺禾致源对二倍体复杂基因组进一步细分为微杂合基因组(%<杂合率<%=、高杂合基因组(杂合率>%)以及高重复基因组(重复序列比例>50%)。 4、怎么查询基因组的大小 答:查询植物基因组大小的网站:; 查询动物基因组大小的网站:。
5、基因组的项目周期 6、基因组承诺的组装指标 答:简单基因组:contig N50>20K,scaffold N50>500K; 复杂基因组:contig N50>20K,scaffold N50>300K。 样品要求 1、动植物基因组测序对取样有什么要求 答:植物:需要黑暗无菌条件下培养的黄化苗、组培苗,基因组样本量500μg~1mg,越多越好。选择纯合或杂合度尽可能小的样品(杂合度<%)。 动物:应选取肌肉、血液等含脂肪较少的部位取样,尽量选择同一个体取样,以减少个体差异性对后续拼接的影响。基因组样本量
500μg~1mg,越多越好。样本的性别决定模式是XY型,则尽量选择雌性个体(XX型),如果是ZW型,则尽量选择雄性个体(ZZ型)。 2、全基因组测序对DNA样本有什么要求 答:(1)样品需求量(单次):小片段文库,≥3μg;2Kb~5Kb大片段文库,≥20μg;10Kb~20Kb大片段文库,≥60μg;完成全基因组测序样品DNA量需求约为500μg~1mg; (2)样品浓度:对于小片段文库,≥50ng/μl,对于2Kb~5Kb 大片段文库,≥150ng/μl;对于10Kb~20Kb大片段文库,≥150ng/μl; (3)样品纯度:OD260/280=~;无蛋白质、RNA污染或肉眼可见杂质污染; (4)样品质量:基因组完整。如需建立≥5Kb的插入片段文库,则电泳结果,基因组DNA主带≥23Kb;脉冲场电泳结果,基因组DNA 主带≥40Kb。 文库构建 1、基因组测序的文库构建及测序策略 答:简单基因组:180bp、500bp、2K、5K、10K;PE100测序;测序深度一般为100-150X;
紫花苜蓿基因组测序及分析 近年来,随着基因组测序技术的不断发展越来越多的植物基因组序列得到完善。通过基因组测序,可以系统地解析植物基因功能。 紫花苜蓿具有优良的农艺性状,是全世界种植范围最广的牧草作物,具有“牧草之王”的美称。在我国,紫花苜蓿生长主要集中在北方地区,这些地区气候寒冷,容易发生低温冻害,造成苜蓿减产,大大降低苜蓿的生产效益。 肇东苜蓿作为紫花苜蓿的地方品种之一,具有良好的抗寒特性。因此,本研究以肇东苜蓿为主要研究材料,进行基因组测序、组装及注释,获得基因组草图;对其它四种紫花苜蓿品种:阿尔冈金、WL168、WL525和WL440进行基因组重测序, 挖掘抗寒相关的SNP。 同时,对紫花苜蓿AP2/ERF转录因子进行挖掘分析,构建紫花苜蓿AP2/ERF 基因调控网络,系统的解析紫花苜蓿AP2/ERF转录因子在抗寒胁迫中的调控机制。主要研究成果如下:1.紫花苜蓿全基因组组装及注释分析通过高通量测序,评估 紫花苜蓿基因组大小为1107M左右,杂合率为1.77%,通过组装获得紫花苜蓿基因组草图。 进一步注释分析,挖掘119194个蛋白质编码基因,它们主要的功能集中在信号转导机制、蛋白质翻译后修饰、防御机制以及转录调控等生物过程。2.紫花苜蓿抗寒性状相关遗传变异的挖掘通过四个紫花苜蓿品种进行基因组重测序,发掘紫花苜蓿品种间的遗传变异(SNP),获得8909604个遗传变异;结合5个紫花苜蓿品种的抗寒性数据,挖掘82838个SNP与紫花苜蓿抗寒性状形成相关。 这些SNP分布在14372个基因上,它们主要涉及转录调控、还原代谢和信号转导等过程。进一步富集分析发现,AP2/ERF、bHLH、MADS、NAC、MYB以及WRKY
首页 科技服务 医学检测 科学与技术 市场与支持 加入我们 关于我们 提供领先的基因组学解决方案Providing Advanced Genomic Solutions 参考文献 [ 1 ] Li Y, Zhou G, Jiang W, et al. De novo assembly of soybean wild relatives for pan-genome analysis of diversity and agronomic traits[J]. Nature biotechnology, 2014, 32(10): 1045-1052. [ 2 ] Da Silva C, Zamperin G, et al. The high polyphenol content of grapevine cultivar tannat berries is conferred primarily by genes that are not shared with the reference genome. Plant Cell, 2013, 25(12):4777-88. [ 3 ] Qi XP, Li M, et al. Identification of a novel salt tolerance gene in wild soybean by whole-genome sequencing. Nature Communica- tions, 2014(5). 挖掘特异基因 解析特有性状 重要品种 全基因组 de novo 所研究品种 小片段文库大片段文库 HiSeq测序(>100X) 基因组组装 注释 全基因组序列比对 转录组遗传图谱等辅助验证 重要农艺性状解析 基因家族聚类分析 所研究品种基因组序列 已发表品种 基因组序列 已发表品种基因集合 所研究品种基因集合 变异检测 小的插入 缺失 SNP 倒位 易位大的插入 缺失 基因挖掘 新基因鉴定拷贝数扩增基因基因丢失正选择基因鉴定 物种 品种 发表杂志(年份) 物种 品种发表杂志(年份) 大豆 棉花 番茄 土豆 水稻 葡萄猪栽培大豆7种野生大豆 野生耐盐大豆雷德氏棉 亚洲棉 陆地棉栽培番茄 抗病番茄栽培土豆 耐寒土豆 Nature (2010) Nature Biotechnology (2014) Nature Communications (2014) Nature (2012)Nature Genetics (2014)Nature Biotechnology (2015)Nature (2012) Nature Genetics (2014) Nature (2011) Plant Cell (2015) 栽培水稻(粳稻)栽培水稻(籼稻) 短花药野生稻 非洲栽培稻五种野生稻三种栽培稻葡萄 丹娜葡萄杜洛克猪藏猪 Science (2002)Science (2002) 野生大豆泛基因组 阅读原文>> 诺禾致源的项目经验 诺禾致源在动植物全基因组测序领域一直处于领先地位, 以第一通讯作者发表基因组文章5篇(影响因子累计152.474),其中2篇为杂志封面文章。 近年来,诺禾通过自主研发软件与技术革新,成功地将项目周期压缩至14个月内,费用降低一半以上。 特有基因检测 对7株代表性野生大豆品种进行全基因组de novo 测序及比较基因组分析,发现每个大豆品种中有1,000~3,000个品种特有的基因。 高变区变异检测 在传统测序方法中,将研究物种短reads 比对参考基因组无法检测到变异位点;在全基因组de novo 方法中,将组装后的超长序列比对参考基因组可准确识别高变区域内的所有变异位点。性状解析方案设计 通过对重要品种高深度(>100X)测序,并进行基因组组装注释: 找到传统测序无法鉴定的高度变异位点,找到更多更准确的SNP位点;找到参考基因组中 所不存在的基因——品种特有基因。 2. 耐盐大豆耐盐基因的发现 2014年,研究人员对一株耐盐大豆开展了全基因组de novo 测序,并与栽培大豆基因组进行全基因组比对,通过一条跨过长达388 Kb 的重要功能区的scaffold,发现了巨大的结构变异,从而成功鉴定出耐盐基因。该基因在栽培大豆中被插入了一个长达3.4 Kb 的反转座子,影响了阳离子转运体功能,从而使栽培大豆失去了耐盐能力。 传统的测序手段,采用的是短reads 比对,因而对这类大的结构变异检测精度差、灵敏度低、甚至难以实现检测,而全基因组de novo 测序则能很好的克服该问题。阅读原文>> 案例分享 1. 丹娜葡萄全基因组测序揭示高丹宁含量性状的分子机制 丹娜葡萄被认为是丹宁含量最高的葡萄品种之一,由于富含丹宁等抗氧化分子,被认为有延缓衰老的作用。通过对其基因组测序,研究人员发现与丹宁合成有关的关键酶,几乎都能找到新的基因。很显然,只依赖已有的参考基因组,完全无法了解丹娜葡萄高丹宁含量这一性状的遗传基础,而全基因组de novo 测序则完美回答了该问题。阅读原文>> 重要品种基因组的价值 一些物种,虽然已有参考基因组,但仍然无法找到性状关联基因。 一方面,参考基因组与研究物种差异太大; 另一方面,性状相关基因处于基因组快速进化区域,变异极大,传统测序手段难以鉴定。 目前,de novo测序在有参品种重要性状探究方面的应用愈发广泛, 相关研究结果常见于国际顶级杂志上。