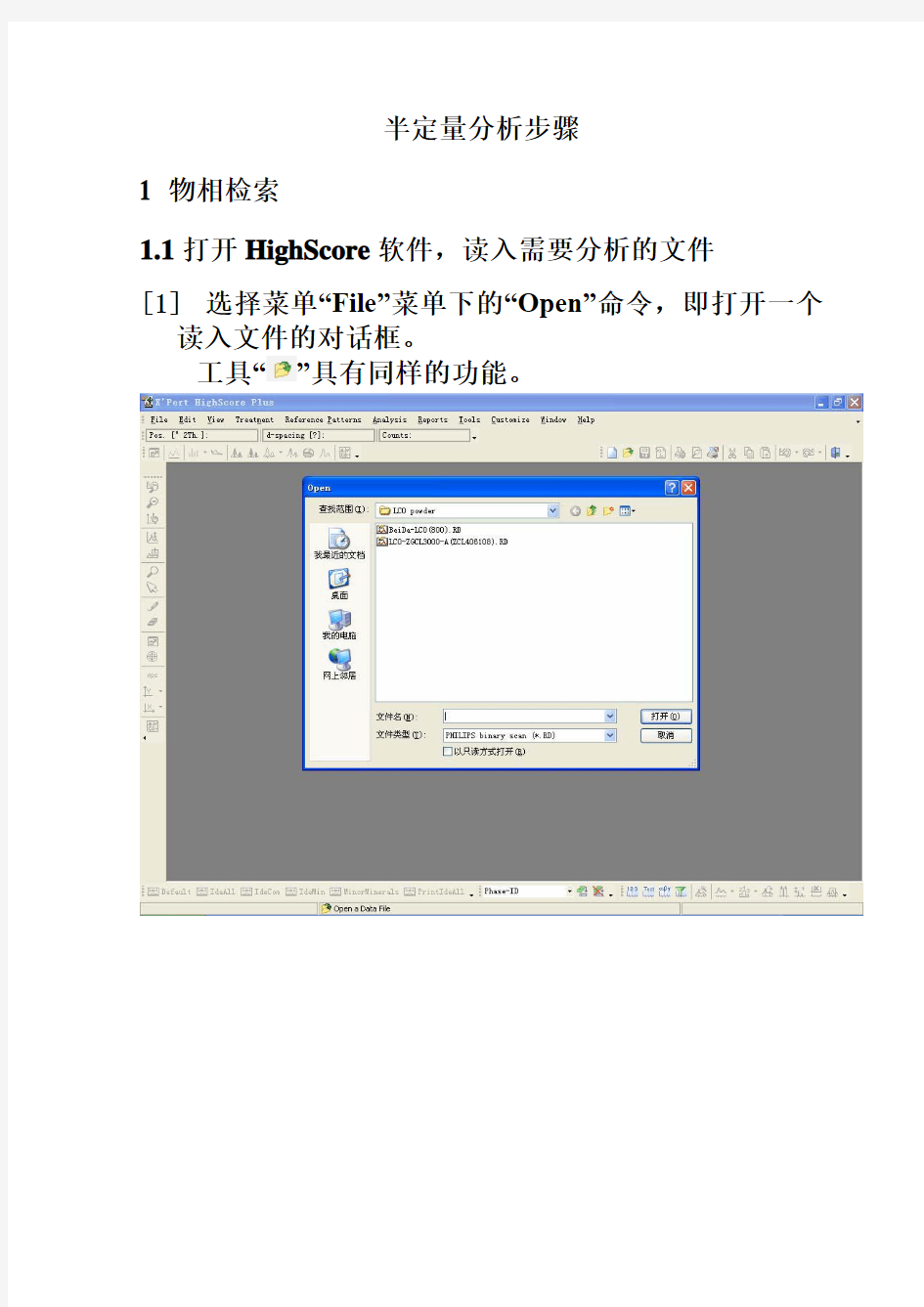

半定量分析步骤
1 物相检索
1.1打开HighScore软件,读入需要分析的文件
[1]选择菜单“File”菜单下的“Open”命令,即打开一个
读入文件的对话框。
工具“”具有同样的功能。
[2] 在文件名上双击,这个文件就被“读入”到主窗口并显示出来。
1.2检索物相
[1] 剔除2
K 峰。
点击菜单栏Treatment→Strip K -Alpha2出现下图所示对话框:
点击strip 2
K ,然后点击replace ,操作完成。
[2] 确定背景
点击菜单栏Treatment→De termine background 出现下图所示对话框:
有三种方式,如果峰形敏锐时一般可以选择自动。手动的方式比较麻烦。如果试样非晶成分较多,会在20-30°之间形成一个非晶包,自动的方式一般会剔除这个非晶包,使用者需要注意。
[3]点击“IdeAll”。
检索结果如下:
[4]选定合适的卡片
检索后,系统会将备选相的卡片列在备选相窗口里。一般备选相中会有很多张卡片待选。系统将卡片和实测衍射谱进行对比,并根据其符合的程度不同给每一张卡片打分,分数高者列在最前面。此时操作者再根据对试样信息的掌握加以甄选。如果对试样的信息一无所知,仅凭得分高低进行判断,出错的几率是很高的。也就是说,最终的评判要由人来完成,而不是机器。
当我们选出第一相后,如果系统在Auto residue(命令菜单为:View→Tool bars→pattern tool bas→ auto residiue)状态下就会自动把剩余的残谱重新进行比对、
打分和排序,方便我们更快的找出第二相、第三相。
点击红圈所示的快捷按钮,或者点击命令菜单:Tools→Reference code即可进入以下界面:
在弹出的对话框里输入已知的PDF 卡号,再点击对话框里的“Load”即可。
注意:(1)主图下方有一个pattern的按钮
在文件刚打开的时候是灰色的,当有卡片选出时,该按钮被激活。按下该按钮,仔细观察实测峰和卡片。如果在实测峰里找不到卡片上的最强峰(100%),无论次强峰对应多么好,都认为试样中没有该卡片上标定的物相。
(2)分析结束后要保存为*.Caf 格式,只有该格式才能保存以上操作所有信息。
2半定量分析
2.1半定量分析原理
从1978年开始,ICDD 发表的PDF 卡片上开始附加有 RIR 值,它是按样品重量与32O Al -α 按1:1 的质量分数混合后,测量样品最强峰的积分强度与刚玉最强峰的积分强度比,如式2-1所示:
col i i I I RIR /= 2-1
其中,Ii 和Icol 分别为物相i 和参考物3
2O Al -α的最强峰
的积分强度。根据“绝热法”原理。如果一个系统中存在N 相,其中第i 相的质量分数可以由各个物相的RIR 值给出,如式2-2[1]:
∑==N i RIRi
Ii RIRi
Ii Wi 1//
2-2
由于影响XRD 强度的因素很多,因而RIR 值的测定也受到合金的晶体结构和化学组成以及实验条件的影响;晶粒的大小、第三组元固溶引起的化学成分改变和粉末颗粒度等因素都会影响RIR 值,而且各物相的衍射峰的峰形、峰宽都不相同,因而利用RIR 值只能做半定量分析。对于精扫且具有很好的峰形结构的XRD 谱线,定量相分析得到的相含量相对误差可以控制在5%左右;对于物相鉴定用连续扫描谱线,半定量分析的物相含量相对误差一般在5%~10%之间[2]。
2.2半定量分析步骤
2.2.1查看物相标准卡片确定物相RIR 值
物相分析完成后,选定物相窗口中的某一组分i 选项双击。
即出现卡片窗口,卡片中的RIR 值即为组分i 的参比强度。
注意:由于种种原因同一个相可能有很多张卡片,而这些卡片所给出的RIR 值不尽相同。操作者再选择卡片时应注意以下标准[3]:
[1] 选择质量标识为S 的卡片(质量标识在卡片Quality 项显示),它可信程度也很高,也经常被引用;再次为I ;随后是C 。
[2] 选择卡片号比较大的,如80 或90 开头的卡片。
这些卡片制做的年代比较晚,手段先进,可信程度相对较高。
[3] 最后还可以对多张卡片的RIR 值进行综合的考虑。比如求个平均值,看哪一张卡片的RIR 最接近平均值就用哪一张。但一般不要用最高或最低的RIR 卡片参与求平均值。
[4] 有些卡片没有RIR 值,这个时候就不能用这个方法了。如果实在需要,就自己查阅一下相关的文献资料找到一个RIR ,或者自己动手求出RIR 。做法也很简单:用纯的、结晶好的本相和刚玉(32O Al -α)混匀研磨、扫描,然后计算各自最强峰的强度,得出RIR 。
[5] 特别的是刚玉(3
2O Al -α)的RIR 值一定要选1。 2.2.2物相最强峰积分强度确定
[1] 查看物相标准卡片确定物相最强峰出现角度。
按上节所述方法打开物相标准卡片。在PEAK LIST(峰列表)一栏找到I%(相对峰强度)为100的峰的出现角度(θ2角)。如下图所示:
[2]在列表上寻找物相最强峰
在选定物相窗口单击peak list按钮,出现谱图所有峰列表。
根据卡片上查得的最强峰θ2角,在峰列表中选择角度最接近的峰为此物相衍射最强峰。
[3]单峰拟合
点击命令菜单View→set manual ranges出现以下界面:
通过在start pos项输入起始角度,在End pos输入终止角度,将最强峰单独选择出来。也可以在谱图上直接通鼠标框选出来最强峰范围。
点击命令菜单Treatmen t→Fit profile进行单峰拟合,或者在谱图上点击鼠标右键选择Fit profile。
[4]记录积分强度
最强峰积分强度就等于峰面积。在Peak list列表里Area(cts*°2Th.)栏目下找到最强峰面积,记录下积分强度Ii。
注意:(1)若同一物相的几个强峰角度接近造成峰位重叠,拟合后要仔细观察图谱检查是否漏峰。如果漏峰需要手动插峰。单击命令菜单Treatment→Insert peak或者在主窗口单击鼠标右键选择Insert peak,在漏峰位置单击鼠标补峰。然后再次拟合记录最强峰面积。
(2)若不同物相最强峰重叠无法辨认,对于无法识别的最强峰,可通过次强峰来估算最强峰面积[1]。打开标准物相卡片,在PEAK LIST表中相对强度值I%一栏找到次强峰的相对强度。
用次强峰强度除以相对强度即为最强峰强度。
2.2.3计算
得到全部物相的RIR 值和最强峰积分强度后根据式2-3即可得到每种物相的质量分数。
∑==N i RIRi Ii RIRi Ii Wi 1// 2-3
参考文献
[1] 夏秀文,张新琴,曹春燕.利用参比强度法确定半定量相分析标定磁性样品金相物相[J].井冈山大学学报(自然科学版),2010,31(5):35-37.
[2] 刘仕.一种实用的X 射线无标定量相分析方法[J].岩石矿物学杂志,1994,13(3):268-277.
[3] 许聚良.Xert Highscore 使用手册.武汉科技大学.
[4]
请将以下内容手写或打印在中原工学院实验报告纸上。 实验报告内容:文中红体字部分请删除后补上自己写的内容班级学号姓名 综合实验X射线衍射仪的使用及物相分析 实验时间,地点 一、实验目的 1.了解x射线衍射仪的构造及使用方法; 2.熟悉x射线衍射仪对样品制备的要求; 3.学会对x射线衍射仪的衍射结果进行简单物相分析。 二、实验原理 (X射线衍射及物相分析原理分别见《材料现代分析方法》第一、二、三、五章。)三、实验设备 Ultima IV型变温全自动组合粉末多晶X射线衍射仪。 (以下为参考内容) X衍射仪由X射线发生器、测角仪、记录仪等几部分组成。
图1 热电子密封式X射线管的示意图 图1是目前常用的热电子密封式X射线管的示意图。阴极由钨丝绕成螺线形,工作时通电至白热状态。由于阴阳极间有几十千伏的电压,故热电子以高速撞击阳极靶面。为防止灯丝氧化并保证电子流稳定,管内抽成1.33×10-9~1.33×10-11的高真空。为使电子束集中,在灯丝外设有聚焦罩。阳极靶由熔点高、导热性好的铜制成,靶面上被一层纯金属。常用的金属材料有Cr,Fe,Co,Ni,Cu,Mo,W等。当高速电子撞击阳极靶面时,便有部分动能转化为X射线,但其中约有99%将转变为热。为了保护阳极靶面,管子工作时需强制冷却。为了使用流水冷却和操作者的安全,应使X射线管的阳极接地,而阴极则由高压电缆加上负高压。x射线管有相当厚的金属管套,使X射线只能从窗口射出。窗口由吸收系数较低的Be片制成。结构分析用X射线管通常有四个对称的窗口,靶面上被电子袭击的范围称为焦点,它是发射X射线的源泉。用螺线形灯丝时,焦点的形状为长方形(面积常为1mm×10mm),此称为实际焦点。窗口位置的设计,使得射出的X射线与靶面成60角(图2),从长方形的短边上的窗口所看到的焦点为1mm2正方形,称点焦点,在长边方向看则得到线焦点。一般的照相多采用点焦点,而线焦点则多用在衍射仪上。 图2 在与靶面成60角的方向上接收X射线束的示意图 自动化衍射仪采用微计算机进行程序的自动控制。图3为日本生产的Ultima IV型变温全自动组合粉末多晶X射线衍射仪工作原理方框图。入射X射线经狭缝照射到多晶试样上,衍射线的单色化可借助于滤波片或单色器。衍射线被探测器所接收,电脉冲经放大后进人脉冲高度分析器。信号脉冲可送至计数率仪,并在记录仪上画出衍射图。脉冲亦可送至计数器(以往称为定标器),经徽处理机进行寻峰、计算峰积分强度或宽度、扣除背底等处理,并在屏幕上显示或通过打印机将所需的图形或数据输出。控制衍射仪的专用微机可通过带编码器的步进电机控制试样(θ)及探测器(2θ)进行连续扫描、阶梯扫描,连动或分别动作等等。目前,衍射仪都配备计算机数据处理系统,使衍射仪的功能进一步扩展,自动化水平更加提高。衍射仪目前已具有采集衍射资料,处理图形数据,查找管理文件以及自动进行物相定性分析等功能。 物相定性分析是X射线衍射分析中最常用的一项测试,衍射仪可自动完成这一过程。首先,仪器按所给定的条件进行衍射数据自动采集,接着进行寻峰处理并自动启动程序。
报告编号:YT-FS-1775-17 XRD物相分析实验报告范 本(完整版) After Completing The T ask According To The Original Plan, A Report Will Be Formed T o Reflect The Basic Situation Encountered, Reveal The Existing Problems And Put Forward Future Ideas. 互惠互利共同繁荣 Mutual Benefit And Common Prosperity
XRD物相分析实验报告范本(完整版) 备注:该报告书文本主要按照原定计划完成任务后形成报告,并反映遇到的基本情况、实际取得的成功和过程中取得的经验教训、揭露存在的问题以及提出今后设想。文档可根据实际情况进行修改和使用。 一、实验目的 1.掌握X 射线衍射仪的使用及进行定性相分析 的基本原理。 2.学会用PDF软件索引对多相物质进行相分析的 方法和步骤。 二、实验原理 布拉格方程:2dsinn X 射线衍射仪是按着晶体对X 射线衍射的几何 原理设计制造的衍射实验仪器。在测试过程,由X 射 线管发射出来的 X 射线照射到试样上产生衍射效应, 满足布拉格方程的2dsinn,和不消光条件的衍射光用 辐射探测器,经测量电路放大处理后,在显示或记录 装置上给出精确的衍射峰位置、强度和线形等衍射信
息,这些衍射信息可作为各种应用问题的原始数据。X 射线衍射仪的基本组成包括;X 射线发生器、衍射测角仪、辐射探测器、测量电路和控制操作、运行软件的电子计算机系统。在衍射测量时,试样绕测角仪中心轴转动,不断地改变入射线与试样表面的夹角,射测量时,试样绕测角仪中心轴转动,不断地改变入射线与试样表面的夹角,与此同时计数器沿测角仪圆运动,接收各衍射角所对应的衍射强度。任何一种结晶物质都具有特定的晶体结构。在一定波长的X 射线照射下,每种晶体物质都产生自己特有的衍射花样。每一种物质与它的衍射花样都是一一对应的,不可能有两种物质给出完全相同的衍射花样。如果试样中存在两种以上不同结构的物质时,每种物质所特有的衍射花样不变,多相试样的 衍射花样只是由它所含各物质的衍射花样机械叠加而成。在进行相分析时,只要和标准的PDF衍射图谱比较就可以确定所检测试样里面的所存在的相。 三、实验仪器,试样
报告编号:LX-FS-A14980 XRD物相分析实验报告标准范本 The Stage T asks Completed According T o The Plan Reflect The Basic Situation In The Work And The Lessons Learned In The Work, So As T o Obtain Further Guidance From The Superior. 编写:_________________________ 审批:_________________________ 时间:________年_____月_____日 A4打印/ 新修订/ 完整/ 内容可编辑
XRD物相分析实验报告标准范本 使用说明:本报告资料适用于按计划完成的阶段任务而进行的,反映工作中的基本情况、工作中取得的经验教训、存在的问题以及今后工作设想的汇报,以取得上级的进一步指导作用。资料内容可按真实状况进行条款调整,套用时请仔细阅读。 一、实验目的 1.掌握X 射线衍射仪的使用及进行定性相分析的基本原理。 2.学会用PDF软件索引对多相物质进行相分析的方法和步骤。 二、实验原理 布拉格方程:2dsinn X 射线衍射仪是按着晶体对X 射线衍射的几何原理设计制造的衍射实验仪器。在测试过程,由X 射线管发射出来的X 射线照射到试样上产生衍射效应,满足布拉格方程的2dsinn,和不消光条件的衍
射光用辐射探测器,经测量电路放大处理后,在显示或记录装置上给出精确的衍射峰位置、强度和线形等衍射信息,这些衍射信息可作为各种应用问题的原始数据。X 射线衍射仪的基本组成包括;X 射线发生器、衍射测角仪、辐射探测器、测量电路和控制操作、运行软件的电子计算机系统。在衍射测量时,试样绕测角仪中心轴转动,不断地改变入射线与试样表面的夹角,射测量时,试样绕测角仪中心轴转动,不断地改变入射线与试样表面的夹角,与此同时计数器沿测角仪圆运动,接收各衍射角所对应的衍射强度。任何一种结晶物质都具有特定的晶体结构。在一定波长的X 射线照射下,每种晶体物质都产生自己特有的衍射花样。每一种物质与它的衍射花样都是一一对应的,不可能有两种物质给出完全相同的衍射花样。如果试样中存在两种以上不同结构的物质时,每种物
关于物相的定量分析 第一个问题:为什么不能做物相定量?样品往往不是单一物相,因此,人们总想了解其中某种相的含量。人们的理解总是认为哪怕只是一种近似的结果,也比没有结果要好。为了要说明定量分析的问题,我们还是了解一下,一张X射线衍射谱图中包含一些什么信息。这些信息主要有三个方面,也是三个方面的应用:一是衍射峰的位置。这方面的信息主要用于物相的鉴定、晶胞参数的精修、残余应力的测 量。二是衍射峰的峰高或者面积,我们称之为强度。这方面的信息主要用于物相的含量、结晶度以及织构的计算。三是衍射峰的形状,我们称为线形。这方面的信息又包括两个方面,其一是衍射峰的宽度,我们可以用来计算亚晶尺寸的大小(常被称为晶粒大小)和微观应变的 计算。另一个则是线的形状,主要是指峰形是否对称,这方面用来计算位错、层错等。不过,后者做的人少,研究也不是很完全,因此,应用不是很广泛。从上面的了解,我们应当知道,不同的实验目的,实验的观察点不同,也就是强调的对象是不同的,如果仅仅为了鉴定物相,一个常规的实验条件就完全可以应付,如果要做晶胞的精修,则需要严格一些的 实验条件。如果要做定量分析,我们的强调点是峰的强度。我们为什么能利用衍射谱来做物相的含量分析呢?其原理就是基于物相的含量W与该物相的衍射强度成正比。可以简单地写成W=CI。W是物相的质量分数,I是该物相的衍射强度。C是一个系数,但不是一个常系数。不过,在一定条件下它是一个常数。遗憾的是,这个常数通常不能通过理论计算得出,而是需要通过实验来测量,每当实验条件改变(包括样品中的物相种
类的改变、任一物相含量的改变、观察峰的改变、甚至于物相产地改变、所用辐射改变、晶粒尺寸改变……如此等等,不一而足)这个系数是变化的。围绕如何想办法得到这个系数C,历代的大师和小师推导出了十几种具体的测量方法,而这些方法又是在某种环境下能使用在另一种环境下不能使用的。每种方法的不同要求等于给实验方法本身加上了一把锁,使得人们不能真正好好地、简便地利用它。这些方法主要包括两方 面:一种是需要标样的,称为“有标法”。也就是说,除了要测的样品,需要往样品中加入某种纯物质。而这些个纯物质往往是不易求得的。比如,某人在合成一种新物质,总是发现合成物中有各种各样的杂质,他希望计算一下不同条件下这种新物质的含量。实验方法要求他提供这种新物质的纯样品。而实际情况是,他如果得到了这种新物质的纯样品,也就是他合成成功的那一天,他还需要你来算个什么含量呢?再举个例子,一位包工头发现,建的房子老是倒了。地基不行。因为地基里有含量很高的蒙脱土,一下雨就膨胀,房子就会倒。他需要了解这种土里的蒙脱土含量到底有多少,他便可以通过改性的办法来解决问题。虽然,要得到蒙脱土的纯物质对某些人来说(一般实验室还是很难的)还是可以的,通过离心等一些方法可以得到。但是,得到的纯物质也许与原样品中的该物质结构发生某些变化。这样虽然得到了纯物质,但是,由于结构的变化,使C也变了。计算出来的结果还是不准。而且,当实验员告诉包工头,虽然我们可以做,但是一则计算结果可能不是很准确,二则经费需要很多时,包工头只能摇摇头,摆摆手,说声B-B了。由于有标法很难用,因此有人就着手研究“无标法”了。这些方法通过理论计算K,或者按某种方法
连续光谱 特征光谱 2 4 6 8 10 α β W Mo Cr 0.5 0.9 0.7 波长(?) 强度 37.2 15.2 图4—1 X 射线管产生的X 射线的波长谱 X 射线衍射晶体结构分析 【摘要】本次实验主要通过采用与X 射线波长数量级接近的物质即晶体这个天然的光栅来作狭缝来研究X 射线衍射,由布拉格公式以及实验中采用的NaCl 晶体的结构特点即可在知道晶格常数条件下测量计算出X 射线的波长,反过来也可用它来测定各种晶体的晶格结构。通过本次实验我们将更进一步地了解X 射线的产生、特点和应用。 【关键词】X 射线;晶体结构;布拉格公式; 1 引言 X 射线是波长介于紫外线和γ射线 间的电磁辐射。由德国物理学家W.K.伦琴于1895 年发现,故又称伦琴射线。波长小于0.1埃的称超硬X 射线,在0.1~1埃范围内的称硬X 射线,1~10埃范围内的称软X 射线。 伦琴射线具有很高的穿透本领,能透过许多对可见光不透明的物质,如墨纸、木料等。这种肉眼看不见的射线可以使很多固体材料发生可见的荧光,使照相底片感光以及空气电离等效应,波长越短的X 射线能量越大,叫做硬X 射线,波长长的X 射线能量较低,称为软X 射线。 实验室中X 射线由X 射线管产生,X 射线管是具有阴极和阳极的真空管,阴极用钨丝制成,通电后可发射热电子,阳极(就称靶极)用高熔点金属制成(一般用钨,用于晶体结构分析的X 射线管还可用铁、铜、镍等材料)。用几万伏至几十万伏的高压加速电子,电子束轰击靶极,X 射线从靶极发出。电子轰击靶极时会产生高温,故靶极必须用水冷却,有时还将靶极设计成转动式的。 目前,X 射线学已渗透到物理学、化学、地学、生物学、天文学、材料科学以及工程科学等许多学科中,并得到了广泛的应用。本实验通过对X 射线衍射实验的研究来进一步认识其性质。 2 实验原理 2.1 X 射线的产生和X 射线的光谱 实验中通常使用X 光管来产生X 射线。在抽成真空的X 光管内,当由热阴极发出的电子经高压电场加速后,高速运动的电子轰击由金属做成的阳极靶时,靶就发射X 射线。发射出的X 射线分为两类:(1)如果被靶阻挡的电子的能量不越过一定限度时,发射的是连续光谱的辐射。这种辐射叫做轫致辐射;(2)当电子的能量超过一定的限时,
实验一 X射线衍射技术及物相分析 一、实验目的与要求 1.学习了解X射线衍射仪的结构和工作原理; 2.掌握X射线衍射物相定性分析的方法和步骤; 3.给定实验样品,设计实验方案,做出正确分析鉴定结果。 二、实验仪器 本实验使用的仪器是Rigaku UltimaⅣX射线衍射仪。主要由冷却循环水系统、X射线衍射仪和计算机控制处理系统三部分组成。X射线衍射仪主要由X射线发生器即X射线管、测角仪、X射线探测器等构成。 1.X射线管 X射线管主要分密闭式和可拆卸式两种。广泛使用的是密闭式,由阴极灯丝、阳极、聚焦罩等组成,功率大部分在1~2千瓦。可拆卸式X射线管又称旋转阳极靶,其功率比密闭式大许多倍,一般为12~60千瓦。常用的X射线靶材有W、Ag、Mo、Ni、Co、Fe、Cr、Cu等。X射线管线焦点为1×10平方毫米,取出角为3~6度。此X射线管为密闭式,功率为2千瓦。X射线靶材为Cu。 选择阳极靶的基本要求:尽可能避免靶材产生的特征X射线激发样品的荧光辐射,以降低衍射花样的背底,使图样清晰。 2.测角仪 测角仪是粉末X射线衍射仪的核心部件,主要由索拉光阑、发散狭缝、接收狭缝、防散射狭缝、样品座及闪烁探测器等组成。 (1)衍射仪一般利用线焦点作为X射线源S。如果采用焦斑尺寸为1×10平方毫米的常规X射线管,出射角6°时,实际有效焦宽为0.1毫米,成为0.1×10平方毫米的线状X射线源。 (2)从S发射的X射线,其水平方向的发散角被第一个狭缝限制之后,照射试样。这个狭缝称为发散狭缝(DS),生产厂供给1/6°、1/2°、1°、2°、4°的发散狭缝和测角仪调整用0.05毫米宽的狭缝。 (3)从试样上衍射的X射线束,在F处聚焦,放在这个位置的第二个狭缝,称为接收狭缝(RS).生产厂供给0.15毫米、0.3毫米、0.6毫米宽的接收狭缝。 (4)第三个狭缝是防止空气散射等非试样散射X射线进入计数管,称为防散射狭缝(SS)。SS和DS配对,生产厂供给与发散狭缝的发射角相同的防散射狭缝。 (5)S1、S2称为索拉狭缝,是由一组等间距相互平行的薄金属片组成,它限制入射X射线和衍射线的垂直方向发散。索拉狭缝装在叫做索拉狭缝盒的框架里。这个框架兼作其他狭缝插座用,即插入DS,
XRD,以及晶体结构的相关基础知识(ZZ) Theory 2009-10-25 17:55:42 阅读355 评论0 字号:大中小 做XRD有什么用途啊,能看出其纯度?还是能看出其中含有某种官能团? X射线照射到物质上将产生散射。晶态物质对X射线产生的相干散射表现为衍射现象,即入射光束出射时光束没有被发散但方向被改变了而其波长保持不变的现象,这是晶态物质特有的现象。 绝大多数固态物质都是晶态或微晶态或准晶态物质,都能产生X射线衍射。晶体微观结构的特征是具有周期性的长程的有序结构。晶体的X射线衍射图是晶体微观结构立体场景的一种物理变换,包含了晶体结构的全部信息。用少量固体粉末或小块样品便可得到其X射线衍射图。 XRD(X射线衍射)是目前研究晶体结构(如原子或离子及其基团的种类和位置分布,晶胞形状和大 小等)最有力的方法。 XRD 特别适用于晶态物质的物相分析。晶态物质组成元素或基团如不相同或其结构有差异,它们的衍射谱图在衍射峰数目、角度位置、相对强度次序以至衍射峰的形状上就显现出差异。因此,通过样品的X射线衍射图与已知的晶态物质的X射线衍射谱图的对比分析便可以完成样品物相组成和结构的定性鉴定;通过对样品衍射强度数据的分析计算,可以完成样品物相组成的定量分析; XRD还可以测定材料中晶粒的大小或其排布取向(材料的织构)...等等,应用面十分普遍、广泛。 目前XRD主要适用于无机物,对于有机物应用较少。 关于XRD的应用,在[技术资料]栏目下有介绍更详细的文章,不妨再深入看看。 如何由XRD图谱确定所做的样品是准晶结构?XRD图谱中非晶、准晶和晶体的结构怎么严格区分? 三者并无严格明晰的分界。 在衍射仪获得的XRD图谱上,如果样品是较好的"晶态"物质,图谱的特征是有若干或许多个一般是彼此独立的很窄的"尖峰"(其半高度处的2θ宽度在0.1°~0.2°左右,这一宽度可以视为由实验条件决定的晶体衍射峰的"最小宽度")。如果这些"峰"明显地变宽,则可以判定样品中的晶体的颗粒尺寸将小于300nm,可以称之为"微晶"。晶体的X射线衍射理论中有一个Scherrer公式,可以根据谱线变宽的量估算晶粒在 该衍射方向上的厚度。 非晶质衍射图的特征是:在整个扫描角度范围内(从2θ 1°~2°开始到几十度)只观察到被散射的X 射线强度的平缓的变化,其间可能有一到几个最大值;开始处因为接近直射光束强度较大,随着角度的增加强度迅速下降,到高角度强度慢慢地趋向仪器的本底值。从Scherrer公式的观点看,这个现象可以视为由于晶粒极限地细小下去而导致晶体的衍射峰极大地宽化、相互重叠而模糊化的结果。晶粒细碎化的极限就是只剩下原子或离子这些粒子间的"近程有序"了,这就是我们所设想的"非晶质"微观结构的场景。非晶质衍射图上的一个最大值相对应的是该非晶质中一种常发生的粒子间距离。
№.5陕西科技大学学报 Oct.2005Vol.23 JOURNALOFSHAANXIUNIVERSITYOFSCIENCE&TECHNOLOGY ?55?3文章编号:1000-5811(2005)05-0055-04 X射线衍射物相定量分析 吴建鹏,杨长安,贺海燕 (陕西科技大学材料科学与工程学院,陕西咸阳712081 ) 摘要:在RigakuD/max22200pc型X,2定量分析 所用的内标曲线和外标曲线,2完全一致,。 关键词:物相定量分析;内标法;中图分类号:O723:A 0引言 X射线衍射物相定量分析已被广泛的应用于材料科学与工程的研究中。X射线衍射物相定量分析有内标法〔1〕、外标法〔2〕、绝热法〔3〕、增量法〔4〕、无标样法〔5,6〕、基体冲洗法〔7〕和全谱拟合法〔8〕等常规分析方法。内标法、绝热法和增量法等都需要在待测样品中加入参考标相并绘制工作曲线,如果样品含有的物相较多,谱线复杂,再加入参考标相时会进一步增加谱线的重叠机会,给定量分析带来困难。基体冲洗法、无标样法和全谱拟合法等分析方法虽然不需要配制一系列内标标准物质和绘制标准工作曲线,但需要烦琐的数学计算,其实际应用也受到了一定限制。外标法虽然不需要在样品中加入参考标相,但需要用纯的待测相物质制作工作曲线,这在实际应用中也是极为不便的。 本研究在RigakuD/max22200pc型X射线衍射仪分析软件的基础上,开发了X射线衍射物相定量分析中最常用的内标法和外标法,并对这两种分析方法进行了实验验证。 1原理 设样品由N个物相组成,采用衍射仪测定时,由Alexander和Klug导出的N相中第J相的衍射强度公式为: IJ=KJ(1) 式中:IJ———试样中J相衍射峰的积分强度;
XRD实验物相定性分析 一、实验目的 1、学习了解X射线衍射仪的结构和工作原理。 2、掌握X射线衍射物相定性分析的原理和实验方法。 3、掌握X射线分析软件Jade5.0和图形分析软件OriginPro的基本操作。
二、实验仪器 D8 Advance型X射线衍射仪 组成:主要由X射线发生器、测角 仪、辐射探测器、记录单元及附件(高 温、低温、织构测定、应力测量、试样 旋转等)等部分组成。 核心部件:测角仪 (1)测角仪 C-计数管;S1、S2-梭拉缝;D-样品;E-支架;K、 L-狭缝光栏;F-接受光栏;G-测角仪圆;H-样品台; O-测角仪中心轴;S-X射线源;M-刻度盘; 图1. 测角仪结构原理图 图2. 测角仪的光路图 X射线源S是由X 射线管靶面上的线状焦斑产生的线状光源。线状光源首
先通过梭拉缝S1,在高度方向上的发散受到限制。随后通过狭缝光栅K,使入射X射线在宽度方向上的发散也受限制。经过S1和K后,X射线将以一定的高度和宽度照射在样品表面,样品中满足布拉格衍射条件的某组晶面将发生衍射。衍射线通过狭缝光栏L、S2和接受光栏F后,以线性进入计数管C,记录X射线的光子数,获得晶面衍射的相对强度,计数管与样品同时转动,且计数管的转动角速度为样品的两倍,这样可以保证入射线与衍射线始终保持2θ夹角,从而使计数管收集到的衍射线是那些与样品表面平行的晶面所产生的。θ角从低到高,计数管从低到高逐一记录各衍射线的光子数,转化为电信号,记录下X射线的相对强度,从而形成 2 — I的关系曲线,即X射线衍射花样。 相对 (2)X射线发生器 图3. X射线产生装置 X 射线管实际上就是一只在高压下工作的真空二极管,它有两个电极:一个是用于发射电子的灯丝,作为阴极,另一个是用于接受电子轰击的靶材,作为阳极,它们被密封在高真空的玻璃或陶瓷外壳内。X射线管提供电部分至少包含有一个使灯丝加热的低压电源和一个给两极施加高电压的高压发生器。当钨丝通过足够的电流使其发生电子云,且有足够的电压(千伏等级)加在阳极和阴极间、使得电子云被拉往阳极。此时电子以高能高速的状态撞击钨靶,高速电子到达靶面,运动突然收到阻止,其动能的一小部分便转化为辐射能,以X射线的形式放出。产生的X射线通过铍窗口射出。 改变灯丝电流的大小可以改变灯丝的温度和电子的发射量,从而改变管电流和X射线强度的大小。改变X光管激发电位或选用不同的靶材可以改变入射X 射线的能量或在不同能量处的强度。 (3)计数器
把数据读入JADE后,我一般不做任何处理就先检索物相,有人喜欢先平滑,但是,平滑的数据会失真,如果不是衍射图特别不好,建议不要先做平滑,当然也不要扣背景,扣Ka2了。这样做的目的,只是为了最真实地找出物相来。在按下S/M后,也会有一个提问,也是问要不要扣背景,也是回答N的。 鼠标的右键按下S/M,建议不要用左键按。右键按下后会出现一个对话框,这里要做的主要是:选择PDF卡片数据库,我一般只选4个库,即无机物,矿物,ICSD无机物和ICSD矿物。当然,如果你做陶瓷,则也应当加上陶瓷库。 第二个要选择的是要不要加入元素过滤器。这是随时改变的。 一个样品第一次检索时,我一般懒得加元素限定条件,也就是不选择元素过滤器,让系统自动在全部卡片上过一遍,看看能否找得到所要的物相。一般都能找出一个两个物相来,选上它。返回到初始窗口。 右键再次按下S/M,打开条件对话框,加入元素限定条件。 加元素绝对不是一古脑儿都加进去,而是有意识,有步骤地加入。检索的过程,我经常说是一个猜数的过程。自己脑子里先有结果,然后再有意识地去找出来,得到验证。 现在很多人做掺杂,做合金的喜欢做大概有10种元素的合金,是否要一次性地把这些元素都加进去呢?肯定是不行的。有些元素,加入量极微,起一些特殊作用,也形成新相,但是,这些相只有通过电子探针或TEM才能找得出来,用XRD是绝对检测不出来的,因此,在加入元素时,要考虑会形成什么样的相,才加入什么样的元素。即使会形成量较多的相,但是,也不要全部加入,一次加入的元素原则上不超过4个,否则就起不到“元素限定”的作用。在的意识地加入某些元素后,再检索,可能会找到另外一些次要相。 在反复几次后,可能还有一些峰没有找到对应的物相,这时,先点一下“画峰按钮”(即求面积的按钮),在一个较强剩余峰下面划一下,即选定这个峰,再S/M,可能会找到与这个峰对应的物相。 在这里要注意两个问题,一是,不能光看是否与这个峰对应,还应当考虑是否有其它峰与所找出的物相谱对应。 二是要不要加入元素限定,可以先不加入元素,大范围地找,如果找不出,再加入元素限定再找。 如果找选的峰找不出新物相来,则换个峰试试。 检索的步骤就是这样,需要反复地去S/M,支摸索。你可以先把可能的相都选上,然后,再一个一个地验证,如果明显矛盾就去掉。如此反复,以至全部峰都找到相应的物相。 如果我们把样品分类,照我的理解,可以分成三大类: 第一类是天然矿物,第二类是人工合成,第三类就是合金。下面我来说说这些样品的特色。照我的理解,天然矿物是最好分析的,因为就那么些种类,而且,天然矿物的数据库特别地全,所有已知矿物都能找到相应的卡片,不存在“新相”的问题。数据库中的数据最成熟。但是,天然矿物也是最容易出现“错判”的样品,特别是粘土类样品,更是这样。有些粘土类矿物,大家都长得差不多(峰位重合),很难根据一个谱线来说这个样品一定就有什么什
《无极材料测试技术》课程作业 作业要求: 对编号01N2009534的样品XRD测试数据进行物相分析,并计算其平均晶粒尺寸大小与晶胞参数。 1.物相分析过程 使用MDI Jade5.0软件对样品XRD测试数据进行分析,以定性分析样品的物相。 1.1.数据的导入 将测试得到的XRD测试数据文件01N2009534.txt直接拖动到Jade软件图标上,导入数据,得到样品XRD衍射图(图1-1)。 图1-1数据导入Jade5.0后得到的XRD图 1.2.初步物相检索 右键点击键,弹出检索对话框,设定初步检索条件:选择所有类型的数据库;检索主物相(Major Phase);不使用限定化学元素检索(Use Chemistry前方框不打钩)(如图1-2所示)。点击“OK”开始检索,得到的检索结果见图1-3。 从初步检索结果可以看出,最可能的物相有四个:CaB5O8(OH)B(OH)3(H2O)3(图1-3)、CaB6O10·5H2O(图1-4a)、Ca2.62Al9.8Si26.2O72H4.56(图1-4b)和C20H20N16O8S4Th(图1-4c)。其中前
三个均为无机物,第四个为有机金属化合物。 从结果分析,由图1-4b、c中可以看出,这两种物相的标准衍射峰没有与样品衍射峰中的最强峰匹配,因此样品中不含有第三、四中物相或者其主晶相不是第三、四种物相。而从图1-3以及图1-4a中可以看出,两种物相的衍射峰与样品的衍射峰几乎都能对上,并且强弱对应良好,因此样品中主晶相可能为CaB5O8(OH)B(OH)3(H2O)3或CaB6O10·5H2O或者两者的混合物。 图1-2 初步物相检索条件设定 图1-3 经过初步检索得到的检索结果
关于XRD物相定量分析 第一个问题:为什么不能做物相定量? 样品往往不是单一物相,因此,人们总想了解其中某种相的含量。人们的理解总是认为哪怕只是一种近似的结果,也比没有结果要好。 为了要说明定量分析的问题,我们还是了解一下,一张X射线衍射谱图中包含一些什么信息。这些信息主要有三个方面,也是三个方面的应用:一是衍射峰的位置。这方面的信息主要用于物相的鉴定、晶胞参数的精修、残余应力的测量。二是衍射峰的峰高或者面积,我们称之为强度。这方面的信息主要用于物相的含量、结晶度以及织构的计算。三是衍射峰的形状,我们称为线形。这方面的信息又包括两个方面,其一是衍射峰的宽度,我们可以用来计算亚晶尺寸的大小(常被称为晶粒大小)和微观应变的计算。另一个则是线的形状,主要是指峰形是否对称,这方面用来计算位错、层错等。不过,后者做的人少,研究也不是很完全,因此,应用不是很广泛。 从上面的了解,我们应当知道,不同的实验目的,实验的观察点不同,也就是强调的对象是不同的,如果仅仅为了鉴定物相,一个常规的实验条件就完全可以应付,如果要做晶胞的精修,则需要严格一些的实验条件。如果要做定量分析,我们的强调点是峰的强度。我们为什么能利用衍射谱来做物相的含量分析呢?其原理就是基于物相的含量W与该物相的衍射强度成正比。可以简单地写成W=CI。W是物
相的质量分数,I是该物相的衍射强度。C是一个系数,但不是一个常系数。不过,在一定条件下它是一个常数。遗憾的是,这个常数通常不能通过理论计算得出,而是需要通过实验来测量,每当实验条件改变(包括样品中的物相种类的改变、任一物相含量的改变、观察峰的改变、甚至于物相产地改变、所用辐射改变、晶粒尺寸改变……如此等等,不一而足)这个系数是变化的。围绕如何想办法得到这个系数C,历代的大师和小师推导出了十几种具体的测量方法,而这些方法又是在某种环境下能使用在另一种环境下不能使用的。每种方法的不同要求等于给实验方法本身加上了一把锁,使得人们不能真正好好地、简便地利用它。这些方法主要包括两方面:一种是需要标样的,称为“有标法”。也就是说,除了要测的样品,需要往样品中加入某种纯物质。而这些个纯物质往往是不易求得的。比如,某人在合成一种新物质,总是发现合成物中有各种各样的杂质,他希望计算一下不同条件下这种新物质的含量。实验方法要求他提供这种新物质的纯样品。而实际情况是,他如果得到了这种新物质的纯样品,也就是他合成成功的那一天,他还需要你来算个什么含量呢?再举个例子,一位包工头发现,建的房子老是倒了。地基不行。因为地基里有含量很高的蒙脱土,一下雨就膨胀,房子就会倒。他需要了解这种土里的蒙脱土含量到底有多少,他便可以通过改性的办法来解决问题。虽然,要得到蒙脱土的纯物质对某些人来说(一般实验室还是很难的)还是可以的,通过离心等一些方法可以得到。但是,得到的纯物质也许与原样品中的该物质结构发生某些变化。这样虽然得到了纯物质,但是,由于结构的变化,使
X射线衍射的物相分析 一、实验目的: (1)熟悉Philips X射线衍射仪的基本结构和工作原理; (2)学会粉末样品的制样及基本的测试过程; (3)掌握利用X射线衍射谱图进行物相分析的方法; 二、实验仪器 (1)制样:未知粉末样品、药匙、酒精(用于擦拭研钵)、研钵、专用进样片; (2)测试:Philips X'pert X射线衍射仪; 三、实验原理 当一束单色x 射线电磁波照射晶体时,晶体中原子周围的电子受x 射线周期变化的电场作用而振动,从而使每个电子都变为发射球面电磁波的次生波源。所发射球面波的频率、与入射的x 射线相一致。基于晶体结构的周期性,晶体中各个电子的散射波可相互干涉而 叠加,称之为相干散射或衍射。 四、实验条件的选择 (1)用于粉末晶体衍射的射线波长一般为0.5~2.5?,本实验中使用的为Cu靶; (2)滤波片选用Ni,因为滤波片是用于吸收Cu的Kβ线,而Ni的吸收限位于Cu的Kα与Kβ之间且靠近Kα线; (3)狭缝参数的选择:在X射线衍射仪的光路中有五个狭缝:梭拉狭缝(两只)、发散狭缝、散射狭缝、接受狭缝。 a. 梭拉狭缝是用来限制X光垂直发散度的,梭拉狭缝发散度的大小对强度和分辨率都有 很大影响,两只狭缝分别位于X光管之后和探测器前。 b. 发散狭缝是用来限制样品表面初级X射线水发散度的,加大狭缝,分辨率降低但强度 增加,可根据实际所需的测试要求进行调解;
c. 散射狭缝用来减少非相干散射及本底等因素造成的背景,提高峰背比,它与发散狭缝配对使用且角度相同; d. 接受狭缝是用来限定进入探测器的X 衍射线的。它位于衍射线的焦点。测量时如果主要为了提高分辨率,应该选择较小的接受狭缝。如果为了提高衍射强度,则应加大接受狭缝。 五、实验操作 1.样品制备: A .测试对于样品粒径的大小并没有严格的要求,但是粒径过大或者不均匀会谱图中锋的相对高度发生变化,导致在对比所得谱图与PDF 标准卡时需要对衍射峰进行大量的排列组合。 B. 测试样品在装入样品板之前必须用毛玻璃将待测表面打磨至完全光滑,并且保证样品的表面与样品板相平。 2.样品扫描 将样品板装入样品台,将防护罩关闭,设定好控制程序,开始扫描,扫描期间面板“shutter open ”指示灯亮起,此时不可以强行打开防护罩,否则会导致仪器强行停止损坏X 光管; 实验中X 光管的高压值设定为4Kv ,电流35mA ;扫描的起始角为10o ,终止角为80o (2θ) 3.结果保存 扫描完成后,当“shutter open ”指示灯熄灭时,确认防护罩解锁后方可打开,取出样品。将数据在highscore 软件中进行处理,软件可以按照要求标示出图中的峰位置,再用软件去除K β线,标示出各个锋的相对高度及d 值,打印结果。 4.利用标准PDF 卡片对未知粉末进行物相分析 将所得的谱图与标准卡片进行对比,有时可能由于峰的相对强度有偏差导致在查找时要对三强线的顺序作出相应调整。d 值的测量受到仪器状态及其他外在因素的影响有一定偏差,这也给查表过程带来了一定难度。 六、实验结果分析 实验中测得未知粉末样品的三强线(three strong lines )分别是 3.34525 ?、4.25729 ?、1.81808 ?,在标准卡片中查找,由于实验条件等因素限制使得测试结果与标准值有一定偏差,最终确定未知样品粉末为二氧化硅,即合成高纯石英( silicon oxide quartz high-synetic ),PDF 编号为89-3433,标准值的三强线分别为3.40 ?、4.34 ?、2.01 ?。 未知粉末的物理性质:白色固体粉末,无特殊光泽,粒径较小,研磨时发现硬度较大,无特殊气味,初步测试不溶于水和酒精溶液。 d 值偏差计算: 0000 3.34525 3.40 100 1.613.40 -?=
第一部分物相分析 1.打开您的数据。File—read… 打开后的界面如图1: 图1 2.很多人说打开数据后要平滑曲线,但是我个人认为还是先不要平滑的好,因为每一次的平滑曲线操作都会造成数据失真。我更倾向于物相分析完毕后,平滑曲线,使得输出的报告更易读。但是,到底要不要在此平滑曲线取决于您自己。平 滑曲线的操作如下: 右击图2中箭头所指按钮,可以进行参数设置,左击就是平滑曲线。
图2 3.物相分析。一般的,物相分析要至少分3轮进行,这样才能把所有的物相找出来。这3轮分别命名为大海捞针、单峰分析、指定元素分析。 首先左击按钮寻峰。 (1)“大海捞针”物相分析:右击图3箭头所指按钮,出现图4所示标签。在General 选项里,首先勾选上左侧的所有的库,去掉右侧所有的对勾,其他设置如图4所示,最后左击ok。 图3
图4 完成上述步骤,出现图5所示界面。显示了矿物名称、化学式、FOM值、PDF-#、RIR等内容。矿物的排序是按FOM值由小到大排列的,FOM值越小,表示存在这种矿物的可能性越大(但不绝对)。当鼠标左击到一个矿物时,在X衍射图谱显示栏会显示蓝色的线,选择与X衍射图谱拟合最好的矿物,然后在矿物名称前面勾选,表示你认为存在此矿物(如图6)。注意:选择矿物时,要尽量选取有RIR值的矿物,否则后面的定量工作将不能继续。 图5 图6
(2)单峰分析:完成大海捞针后,可能还有峰没有对上,此时要用此法。 在大海捞针的基础上,左击图7方框内的按钮,然后按照图8内标明的步骤操作。然后重复大海捞针的操作(与大海捞针不同的是,此时系统只选择与你选中的峰对应的物相)。 图7
半定量分析步骤 1 物相检索 1.1打开HighScore软件,读入需要分析的文件 [1]选择菜单“File”菜单下的“Open”命令,即打开一个读入文件的对话框。 工具“”具有同样的功能。
[2] 在文件名上双击,这个文件就被“读入”到主窗口并显示出来。 1.2检索物相 [1] 剔除2 K 峰。
点击菜单栏Treatment→Strip K -Alpha2出现下图所示对话框: 点击strip 2 K ,然后点击replace ,操作完成。 [2] 确定背景 点击菜单栏Treatment→De termine background 出现下图所示对话框: 有三种方式,如果峰形敏锐时一般可以选择自动。手动的方式比较麻烦。如果试样非晶成分较多,会在20-30°之间形成一个非晶包,自动的方式一般会剔除这个非晶包,使用者需要注意。 [3] 点击“IdeAll”。
检索结果如下: [4]选定合适的卡片 检索后,系统会将备选相的卡片列在备选相窗口里。一般备选相中会有很多张卡片待选。系统将卡片和实测衍射谱进行对比,并根据其符合的程度不同给每一张卡片打分,分数高者列在最前面。此时操作者再根据对试样信息的掌握加以甄选。如果对试样的信息一无所知,仅凭得分高低进行判断,出错的几率是很高的。也就是说,最终的评判要由人来完成,而不是机器。 当我们选出第一相后,如果系统在Auto residue(命令菜单为:View→Tool bars→pattern tool bas→ auto residiue)状态下就会自动把剩余的残谱重新进行比对、打分和排序,方便我们更快的找出第二相、第三相。
结构分析唐老师部分作业汇总 第一次作业 1、请写出晶体的定义。试说明什么是单晶体?什么是多晶体? 定义:质点(原子、离子或分子)在空间按一定规律周期性重复排列构成的固体物质。基本为一个空间点阵所贯穿的整块固体称单晶体,简称单晶;由许多小单晶按不同取向聚集形成的固体称多晶。 2、晶格与点阵是何关系?晶体结构与点阵、结构基元是何关系?原子参数与阵点坐标是何关系? 晶体是由原子、离子或分子在空间按一定规律周期性重复地排列所构成的固体物质,将其中周期性排列的重复单元抽象成在空间以同样周期性排列的相同几何点,这些点所构成的阵列称为点阵(lattice),或空间点阵、空间格子。沿三个不同的方向,通过点阵中的点阵点可以作许多平行的直线族和平行的晶面族,使点阵形成三维网格。这些将点阵点全部包括在其中的网格称为晶格。带有原子、离子、分子或其集团的点阵就是晶格。 晶体结构= 点阵+ 结构基元 对于点阵点坐标和原子参数,它们对于3个坐标轴的方向是相同的,但是点阵点坐标的度量单位是点阵周期,而原子参数的度量单位是晶胞参数。 3、晶体的晶胞类型共分为哪几种?空间格子(点阵)可分为几类?每一类晶系各有多少种空间点阵格子形式?请分别写出。 晶胞是描述晶体微观结构的基本单元,有素晶胞和复晶胞之分。 如果点阵点都处于平行六面体的顶点,每个平行六面体只有一个点阵点,此空间格子称为素格子,以P表示;如果体心还有点阵点,则此空间格子称为体心格子,以I表示;如果所有平面格子中心有点阵点,则称为面心格子,以F表示;如果仅一对相对的平面格子中心有点阵点,则此空间格子称为底心格子,视相对面位置分别以A, B或C表示。 晶体分为7个晶系(立方、六方、四方、三方、正交、单斜和三斜),依据特征对称元素和正当点阵单位的划分规则,晶体的点阵分为14种空间点阵型式:简立
关于xrd物相分析实验报告范文 篇一:XRD物相分析实验报告 一、实验目的 1.掌握X 射线衍射仪的使用及进行定性相分析的基本原理。 2.学会用PDF软件索引对多相物质进行相分析的方法和步骤。 二、实验原理 布拉格方程:2dsinn X 射线衍射仪是按着晶体对 X 射线衍射的几何原理设计制造的衍射实验仪器。在测试过程,由X 射线管发射出来的 X 射线照射到试样上产生衍射效应,满足布拉格方程的2dsinn,和不消光条件的衍射光用辐射探测器,经测量电路放大处理后,在显示或记录装置上给出精确的衍射峰位置、强度和线形等衍射信息,这些衍射信息可作为各种应用问题的原始数据。X 射线衍射仪的基本组成包括;X 射线发生器、衍射测角仪、辐射探测器、测量电路和控制操作、运行软件的电子计算机系统。在衍射测量时,试样绕测角仪中心轴转动,不断地改变入射线与试样表面的夹角,射测量时,试样绕测角仪中心轴转动,不断地改变入射线与试样表面的夹角,与此同时计数器沿测角仪圆运动,接收各衍射角所对应的衍射强度。任何一种结晶物质都具有特定的晶体结
构。在一定波长的X 射线照射下,每种晶体物质都产生自己特有的衍射花样。每一种物质与它的衍射花样都是一一对应的,不可能有两种物质给出完全相同的衍射花样。如果试样中存在两种以上不同结构的物质时,每种物质所特有的衍射花样不变,多相试样的 衍射花样只是由它所含各物质的衍射花样机械叠加而成。在进行相分析时,只要和标准的PDF衍射图谱比较就可以确定所检测试样里面的所存在的相。 三、实验仪器,试样 XRD仪器为:Philip X’Pert diffractometer with Cu-Ka radiation source (=1.54056) at 40Kv。 实验试样:Ti98Co2基的合金 四、实验条件 2=20-80o step size:0.05o/S 五、实验步骤 1.开总电源 2.开电脑,开循环水 3.安装试样,设置参数,并运行Xray衍射仪。 4.Xray衍射在电脑上生成数据,保存数据。 5.利用orgin软件生成Xray衍射图谱。并依次找出峰值的,并与PDF中的标准图谱相比较,比对三强线的,确定试样中
第一部分物相分析 1.打开您的数据。… 打开后的界面如图1: 图1 2.很多人说打开数据后要平滑曲线,但是我个人认为还是先不要平滑的好,因为每一次的平滑曲线操作都会造成数据失真。我更倾向于物相分析完毕后,平滑曲线,使得输出的报告更易读。但是,到底要不要在此平滑曲线取决于您自己。平 滑曲线的操作如下: 右击图2中箭头所指按钮,可以进行参数设置,左击就是平滑曲线。
图2 3.物相分析。一般的,物相分析要至少分3轮进行,这样才能把所有的物相找出来。这3轮分别命名为大海捞针、单峰分析、指定元素分析。 首先左击按钮寻峰。 (1)“大海捞针”物相分析:右击图3箭头所指按钮,出现图4所示标签。在General 选项里,首先勾选上左侧的所有的库,去掉右侧所有的对勾,其他设置如图4所示,最后左击ok。 图3
图4 完成上述步骤,出现图5所示界面。显示了矿物名称、化学式、FOM值、PDF-#、RIR等内容。矿物的排序是按FOM值由小到大排列的,FOM值越小,表示存在这种矿物的可能性越大(但不绝对)。当鼠标左击到一个矿物时,在X衍射图谱显示栏会显示蓝色的线,选择与X衍射图谱拟合最好的矿物,然后在矿物名称前面勾选,表示你认为存在此矿物(如图6)。注意:选择矿物时,要尽量选取有RIR值的矿物,否则后面的定量工作将不能继续。 图5 图6
(2)单峰分析:完成大海捞针后,可能还有峰没有对上,此时要用此法。 在大海捞针的基础上,左击图7方框内的按钮,然后按照图8内标明的步骤操作。然后重复大海捞针的操作(与大海捞针不同的是,此时系统只选择与你选中的峰对应的物相)。 图7