第3章铁基块体非晶合金-纳米晶转变的动力学模拟过程3.1 Discover模块动力学模拟
3.1.1 原子力场的分配
在使用Discover模块建立基于力场的计算中,涉及几个步骤。主要有:选择力场、指定原子类型、计算或指定电荷、选择non-bond cutoffs。
在这些步骤中,指定原子类型和计算电荷一般是自动执行的。然而,在某些情形下需要手动指定原子类型。原子定型使用预定义的规则对结构中的每个原子指定原子类型。在为特定的系统确定能量和力时,定型原子使工作者能使用正确的力场参数。通常,原子定型由Discover使用定型引擎的基本规则来自动执行,所以不需要手动原子定型。然而,在特殊情形下,人们不得不手动的定型原子,以确保它们被正确地设置。
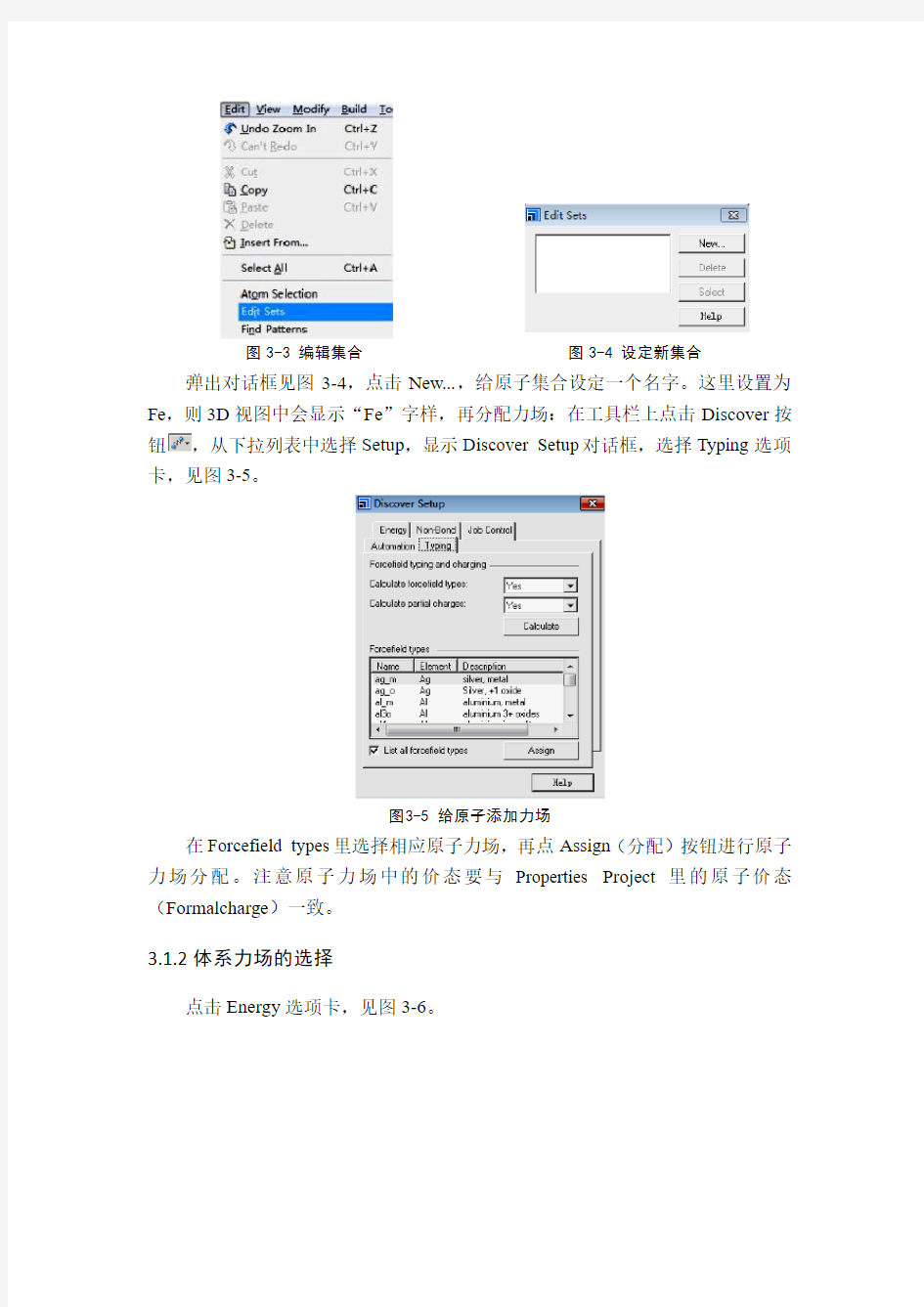
图 3-1调出选择原子窗口图3-2 选择原子窗口计算并显示原子类型:点击Edit→Atom Selection,如图3-1所示。弹出对话框,如图3-2所示。从右边的…的元素周期表中选择Fe,再点Select,此时所建晶胞中所有Fe原子都将被选中,原子被红色线圈住即表示原子被选中。再编辑集合,点击Edit→Edit Sets,如图3-3、3-4所示。
图3-3 编辑集合图3-4 设定新集合弹出对话框见图3-4,点击New...,给原子集合设定一个名字。这里设置为Fe,则3D视图中会显示“Fe”字样,再分配力场:在工具栏上点击Discover按钮,从下拉列表中选择Setup,显示Discover Setup对话框,选择Typing选项卡,见图3-5。
图3-5 给原子添加力场
在Forcefield types里选择相应原子力场,再点Assign(分配)按钮进行原子力场分配。注意原子力场中的价态要与Properties Project里的原子价态(Formalcharge)一致。
3.1.2体系力场的选择
点击Energy选项卡,见图3-6。
图3-6 Energy选项卡图3-7 力场下拉菜单力场的选择:
力场是经典模拟计算的核心,因为它代表着结构中每种类型的原子与围绕着它的原子是如何相互作用的。对系统中的每个原子,力场类型都被指定了,它描述了原子的局部环境。力场包括描述属性的不同的信息,如平衡键长度和力场类型对之间的电子相互作用。常见力场有COMPASS、CVFF和PCFF。
Select下拉菜单中有三个选项,见图3-7:
①COMPASS 力场:COMPASS 力场是第一个把以往分别处理的有机分子体系的力场与无机分子体系的力场统一的分子力场。COMPASS 力场能够模拟小分子与高分子,一些金属离子、金属氧化物与金属。在处理有机与无机体系时,采用分类别处理的方式,不同的体系采用不同的模型,即使对于两类体系的混合,仍然能够采用合理的模型描述。本文采用此力场。
②CVFF力场:CVFF 力场全名为一致性价力场(consistant valence force field),最初以生化分子为主,适应于计算氨基酸、水及含各种官能团的分子体系。其后,经过不断的强化,CVFF 力场可适用于计算多肽、蛋白质与大量的有机分子。此力场以计算系统的结构与结合能最为准确,亦可提供合理的构型能与振动频率。
③PCFF力场:PCFF为一致性力场,增加一些金属元素的力参数,可以模拟含有相应原子的分子体系,其参数的确定除大量的实验数据外,还需要大量的量子力学计算结果[16]。
3.1.3 非键的设置
打开Non-bond选项卡,见图3-8。
图3-8 非键选项卡
非键作用力包括范德华力和库伦力。这里将两者都选上,为的是后期做minimizer优化原子位置时精确度更高,因为考虑的作用力因素多,即两者都考虑了。
Summation method(模拟方法):
①Atom Based:atom based基于原子的总量,包括一个原子的截断距离,一个原子的缓冲宽度距离;为直接计算法,即直接计算原子对之间的非键相互作用,当原子对超出一定距离(截断半径cutoff distance)时,即认为原子对之间相互作用为零(注:cutoff distance指范德瓦尔斯作用力和库仑力的范围,比如:设定截断半径为5,则表示已分子或原子中心为圆心,以5为半径作圆,半径以外的作用力都不考虑)。此方法计算量较小,但是可能导致能量和其导数的不连续性。当原子对间距离在Cutoff半径附近变化时,由于前一步考虑了原子对之间的相互作用,而后一步不考虑,由此会导致能量发生跳跃。当然,对于较小的体系,则可以设置足够大的Cutoff半径来保证所有的相互作用都被考虑进来。见图3-8。
图3-9 非键图
②Group Based:group based基于电子群的,总量中包括一个原子的截断距
离,一个原子的缓冲宽度距离;大多数的分子力场都包括了每个原子之间点电荷的库仑相互作用。甚至在电中性的物种中也存在点电荷,例如水分子。点电荷实际上反映了分子中不同原子的电负性。在模拟中,点电荷一般是通过电荷平衡法(charge equilibrium)评价或者力场定义的电荷来分配的。当评价点电荷时,一定要小心不要在使用Cutoff技术时引入错误的单极项。要了解到这一点,可以参看如下事实:两个单极,当只有1e.u.电荷时,在10A的位置上其相互作用大约为33Kcal;而对于由单位单极分离1A所形成的两个偶极,相同距离其相互作用能不超过0.3Kcal/mol。
很明显,忽略单极-单极相互作用会导致错误的结果,而忽略偶极-偶极相互作用则是适度的近似。然而,如果单极相互作用处理不清的话,仍然会出问题。当non-bond Cutoff使用基于原子-原子基组时,就可能发生,会人为将偶极劈裂为两个“假”的单极(当一个偶极原子在Cutoff内,另一个在其外)。这就不是忽略了相对较小的偶极-偶极相互作用,而是人为引入了作用较大的单极-单极相互作用。为了避免这种人为现象,Materials Studio引入了在Charge Groups之上的Cutoff。
一个“Charge Group”是一个小的原子基团,其原子彼此接近,净电荷为0或者接近于0。在实际应用中,Charge Group一般是常见的化学官能团,例如羰基、甲基或者羧酸基团的净电荷接近于中性Charge Group。Charge Group之间的距离为一个官能团中心到另一个官能团中心的距离R,Cutoff设置与Atom Based相类似。
③Ewald Summation:Ewald是在周期性系统内计算Non-bond的一种技术。Ewald是计算长程静电相互作用能的一种算法。Ewald加和方法比较合适于结晶固体。原因在于无限的晶格内,Cutoff方法会产生较大的误差。然而,此方法放也可以用于无定形固体和溶液体系。Ewald计算量较大,为o(N^3/2),体系较大时,会占用较多的内存并花费较长的时间[19]。
④cell multipole cell based:只能用于基于指定数量层。
一般情况下,基于Atom适合于孤立体系,对于周期性体系计算量较小,但是准确性较差;基于Group适合于周期性和非周期性体系,计算的准确性好一些,计算量最小;Ewald适合于周期性能体系,计算最为准确,但计算量最大。
图3-10 Atom Based Cutoff窗口
本次模拟选择Atom Based模拟方法,弹出对话框,见图3-10。
Cutoff distance(截断距离):指的是范德瓦尔斯作用力和库仑力的范围,见图3-9。
Buffer width:缓冲宽度距离。
Setup其他选项保留默认设置即可。
3.1.4 结构优化
在工具栏上点击Discover按钮,然后选择Minimizer。或者从菜单栏选择Modules | Discover | Minimizer,见图3-11。显示Discover Minimizer对话框,可以进行几何结构优化计算。优化前(Minimizer),先查看所有原子是否都已分配力场,如果没有,可以手动添加,在Properties Explorer中双击Forcefield type,然后修改力场类型即可。其次在Min之前,需要把晶体结构所有原子重新固定。minimizer只是对结构进行优化,以达到能量最小化。在作动力学(dynamics)之前最好执行minimizer,因为如果不执行minimizer,则计算收敛时间会比较长,能量波动会比较大,而且计算有可能出错。
图3-11 几何优化窗口
优化方法Method:最陡下降法(Steepest Descent)、共轭梯度法(Conjugate Gradient)、牛顿方法(Newton)和综合法(Smart Minimizer)。
Convergence level:收敛精度水平。
Maximum iteration:最大迭代数。
Optimize cell选中的话表示优化晶胞参数和原子位置。
MS Discover 结构优化原理
分子的势能一般为键合(键长、键角、二面角、扭转角等)和非键合相互作用(静电作用、范德华作用等)能量项的加和,总势能是各类势能之和,如下式:总势能= 范德华非键结势能+ 键伸缩势能+ 键角弯曲势能+ 双面角扭曲势能+ 离平面振动势能+ 库伦静电势能+ ...
除了一些简单的分子以外,大多数的势能是分子中一些复杂形势的势能的组合。势能为分子中原子坐标的函数,由原子不同的坐标所得到的势能构成势能面(Potential Energy Surface,PES)。势能越低,构象越稳定,在系统中出现的机率越大;反之,势能越高,构象越不稳定,在系统中出现的机率越小。通常势能面可得到许多极小值的位置,其中对应于最低能量的点称为全局最小值(Global Energy Minimum),相当于分子最稳定的构象。由势能面求最低极小值的过程称为能量最小化(Energy Minimum),其所对应的结构为最优化结构(Optimized Structure),能量最小化过程,亦是结构优化的过程。
通过最小化算法进行结构优化时,应避免陷入局部最小值(local minimum),也就是避免仅得到某一构象附近的相对稳定的构象,而力求得到全局最小值,即实现全局优化。分子力学的最小化算法能较快进行能量优化,但它的局限性在于易陷入局部势阱,求得的往往是局部最小值,而要寻求全局最小值只能采用系统搜寻法或分子动力学法。在Materials Studio的Discover模块中,能量最小化算法有以下四种:
①最陡下降法(Steepest Descent),为一经典的方法,通过迭代求导,对多变量的非线性目标函数极小化,按能量梯度相反的方向对坐标添加位移,即能量函数的负梯度方向是目标函数最陡下降的方向,所以称为最陡下降法。此法计算简单,速度快,但在极小值附近收敛性不够好,造成移动方向正交。最陡下降法适用于优化的最初阶段。
②共轭梯度法(Conjugate Gradient),在求导时,目标函数下降方向不是仅选取最陡下降法所采用的能量函数的负梯度方向,而是选取两个共轭梯度方向,即前次迭代时的能量函数负梯度方向与当前迭代时的能量函数负梯度方向的线性组合。此法收敛性较好,但对分子起始结构要求较高,因此常与最陡下降法联合使用,先用最陡下降法优化,再用共轭梯度法优化至收敛。
③牛顿方法(Newton),以二阶导数方法求得极小值。此法的收敛很迅速,也常与最陡下降法联合使用。
④综合法(Smart Minimizer),该方法可以混合最陡下降法,共轭梯度法和牛顿法进行结构优化,在MS中是可选择的。Smart Minimizer中,牛顿法可以设定最大的原子数,如果体系的原子数大于所设定的值,则计算是会自动地转为前面设定的收敛法(共轭梯度法或最陡下降法),收敛精度会改为共轭梯度法的默
认收敛精度值。
点开各种方法后面的More,见图3-12、3-13、3-14、3-15,可设定收敛精度(Convergence),算法(Algorithm)和一维搜索(Line search,指每一次迭代中的精度)等。
图3-12 综合法窗口
图3-13 最陡下降法窗口图3-14 共轭梯度法窗口图3-15 牛顿法窗口
当Job结束后,结果被返回到Disco Min目录,最小化的结构被命名为3D Atomistic.xsd,并被保存在“3D Atomistic Disco Min”目录。还生成一个名为“3D Atomistic.out”的文本文档,它包含了有关计算的所有能量信息。同时还生成“SimulationEnergies.xcd”,它显示了能量随迭代次数的变化情况,由于优化是使结构更加稳定,所以能量也随之降低,最终趋于某一值,如图3-16所示。本次模拟结果前后对比见图3-17,
图3-16 能量-迭代次数函数图
(a)几何优化前(二维)(b)几何优化后(二维)
(c)几何优化前(三维)(d)几何优化后(三维)
图3-17 结构优化前后对比
结构优化后,原子会有所重排,使结构更加稳定。
3.1.5 高温弛豫
打开discover下的Dynamics,如图3-18所示。
图3-18 Dynamics窗口
Ensemble(系综):NVE、NVT、NPT、NPH。
Temperature:目标温度。
Pressure:给系统所施加的压力。
Number of steps:整个动力学所运行的总步数。
Time step:每一动力学步骤所花费的时间(单步长时间)。
Dynamics time:Number of steps × Time step(总模拟时间)。
Trajectory Save:Coordinates表示保存坐标;Final Structure表示只保存最终结构;Full表示保存所有。
Frame output every:若输入5000,则表示每5000步输出一帧(即晶体结构)。
运行结束后,可以通过调用Animation观看三维动画,见图3-19、3-20。
图3-19 调出Animation方式
图3-20 Animation操作图
动画工具条可以控制三维窗口中动画文件的显示,见图3-20。它包含以下命令:
Play Backwards:倒映动画文件。
Step Backwards:每次向后放一帧。
Stop:停止放映。
Step Forwards:每次一帧加速放映。
Play:放映动画。
Pause:暂停放映,再按一次后继续放映。
Animation Mode:显示动画模式下拉菜单。
3.1.6.1 系综简介
系综(ensemble)是指具有相同条件系统(system)的集合。平衡态的分子动力学模拟,总是在一定的系综下进行。系综是统计力学中非常重要的概念,系统的一切统计特性基本都是以系综为起点推导得到的。实际应用时,要注意选择适当的系综,如(N,T,P)常用于研究材质的相变化等。
①NVE(微正则系综)
在微正则系综(micrononical ensemble)中,模型体系的粒子数N、体积V 及内能(热力学能)E(在热力学通常用U表示内能)保持不变。是一个孤立、保守的系统。值得注意的是:体系总能量,即势能和动能的总和,是保持守恒的,
常被用来判断积分的精度固定不变。它对应于绝热过程,即体系与环境没有热交换,不存在温度T和压力p的控制因素。由于体系的能量E是守恒的,体系的动能和势能之间互转化。一般说,给定能量的精确初始条件是无法得到的。能量的调整通过对速度的标度进行,这种标度可能使系统失去平衡,迭代弛豫达到平衡。
②NVT系综(正则系综)
正则系综(canonical ensemble)中,体系的粒子数N、体积V及温度T保持不变,且总动量保持不变。因此正则系综动力学有时也被称为恒温动力学。为了控制体系的温度,就需要设置一个“虚拟”的热浴环境,与体系进行能量交换。常用的热浴(bath)包括:Nose-Hoover,Berendsen,Andersen以及“velocity scaling (速度标定)”方法等。
③NPT系综(恒温恒压系综)
恒温恒压系综中,体系的粒子数N、压力P、温度T都是恒定不变的。恒温恒压系综允许体系的“体积”发生变化。这里的体积的变化有两种方式,一种是只变化尺寸而保持形状(比如对于晶体来说,晶格类型维持不变,但是晶胞参数中的a,b,c可以变化),另一种是同时变化形状和尺寸(即晶格类型和晶胞参数都可以变化)。压强P与体积共轭,控压可以通过标度系统的体积来实现。目前有许多调压的方法都是采用的这个原理。
④NPH系综(恒焓恒压系综)
NPH系综中体系的粒子数N、压力P及体系的焓H(H=E+pV)是守恒的,例如节流膨胀就是一恒焓过程。在模拟中较少见[17]。
点击More…出现如图3-21所示对话框,Energy deviation表示每一步所允许的最大能量偏离为5000kcal/mol。
图3-21 能量偏离设定
NPT系综最符合实验条件,但是在NPT系综下跑动力学过程中晶胞密度变化太大,不符合实际情况,因此本文采用NVT系综。
3.1.6.2 系综控温机制
系综的控温:温度调控机制可以使系统的温度维持在给定值,也可以根据外界环境的温度使系统温度发生涨落。一个合理的温控机制能够产生正确的统计系综,即调温后各粒子位形发生的概率可以满足统计力学法则。系综控温机制主要
有:Velocity Scale、Nose、Berendsen。
图3-22 控温机制的设定图3-23 几种控温机制图3-22的Thermostat下拉菜单有四个,见图3-23:
①V elocity Scale(直接速度标定法):系统温度和粒子的速度直接相关,可以通过调整粒子的速度使系统温度维持在目标值。实际分子动力学模拟中,并不需要对每一步的速度都进行标定,而是每隔一定的积分步,对速度进行周期性的标定,从而使系统温度在目标值附近小幅波动。直接速度标定法的优点是原理简单,易于程序编制。缺点是模拟系统无法和任何一个统计力学的系综对应起来;突然的速度标定引起体系能量的突然改变,致使模拟系统和真实结构的平衡态相差较远。
②Nose:该方法可以把任何数量的原子与一个热浴耦合起来,可以消除局域的相关运动,而且可以模拟宏观系统的温度涨落现象。
③Andersen:体系与一强加了指定温度的热浴相耦合。
④Berendsen控温机制: 又称Berendsen外部热浴法。其基本思想是假设系统和一个恒温的外部热浴耦合在一起,通过热浴吸收和释放能量来调节系统的温度,使之与恒温热浴保持一致。对速度每一步进行标定,以保持温度的变化率与热浴和系统的温差(T bath-T(t))成比例。
当系综选定NPT时,控温机制应用Nose,因此本文采用Nose控温机制。
3.1.6.3 系综控压机制
图3-24 几种控压机制
图3-24下拉菜单有3项:
①Andersen:假定系统与外界“活塞”耦合,当外部压强不能补偿系统内部压强时,“活塞”运动引起系统均匀地膨胀或收缩,最终使得系统压强等于外部
压强。Andersen方法具有重要的意义,后来的各种压力控制方法基本都是基于Andersen思想发展起来的。
②Berendsen:这种方法是假想把系统与一“压浴”相耦合。
③Parrinello:这种方法允许原胞的形状与体积同时发生变化,以达到与外压平衡。这种方法是对Anderson调压方法的一种扩展,可以实现对原胞施加拉伸剪切以及混合加载情况的模拟,因此在对材料的力学性质的分子动力学模拟中,得到了广泛地应用。
由于本文采用NPT系综,压力一定,所以将会看到控压机制不可选。Dynamics运行结束后,所得结构如图3-25所示。
图3-25 高温弛豫后的3D图
由于高温(2000K)弛豫,高于熔化温度,所以此时体系处于液态状态,因此原子处于远程无序状态。
Project Explorer中还生成了其他一些文件,如能量-模拟时间曲线图,见图3-26。温度-模拟时间曲线图,见图3-27。以及一些输入输出文件。
图3-26
图3-27
高温弛豫中原子通过迁移、运动或者扩散,逐步降低原来的高内能态,向稳定的低内能态转变。因此能量随时间的推移将降低并趋于某一值,而温度逐渐稳定在设定的2000K上下做微小变动。
3.2 Forcite模块动力学模拟
3.2.1 Quench(快冷)
在工具栏上点击按钮,选择calculation,弹出对话框,如图3-28所示。
图3-28快冷窗口图3-29快冷参数设置选择Quench(快冷,淬火),再点击More…出现如图3-29所示对话框:再点击Dynamics options的more…出现如图3-30、3-31所示:
图3-30快冷动力学窗口图3-31 速度下拉菜单Initial velocities:第一次由于设置速度,所以只能选择Random(随即速度),第二次以及以后运行则可选择Current(当前速度)了,此时速度为上一次结束的速度。
图3-32
注意:模拟退火的时候要加力。即Include forces要选上,如图3-32所示。
运行结束后会得到一些文件,有1)3D Atomistic.xtd,这是快冷后得到的结构,见图3-33(a)和(b);2)Status.txt以及3D Atomistic.txt包含了快冷过程的相关参数设置以及结果数据;3)3D Atomistic Temperature.xcd描述了温度与时间的关系,见图3-34;4)3D Atomistic Energies.xcd描述了几种能量(势能、动能、非键能以及总能量)随时间的变化关系(见图3-35)等。
(a)二维(b)三维
图3-33 快冷所得结构
图3-34 快冷后的温度-时间曲线
图3-35 快冷后的能量-时间曲线
对图3-33所示的结果做径向分布函数分析,得到如图3-36的图像,表明快冷结果得到非晶合金。
图3-36 径向分布函数
3.2.2 anneal(退火)
选择退火(anneal)如图3-37所示。
图3-37 退火窗口图3-38 退火动力学窗口点击more…出现图3-38所示对话框:
Annealing cycles:运行一次退火所作的退火循环次数。
Initial temperature:一次退火循环的起始温度也是退火循环的终止温度。
Mid-cycle temperature:一次退火循环包括升温过程和降温过程中的最高温度。
Heating ramps per cycle:一次循环中加热过程的温度梯度步数,冷却过程的温度下降梯度(cooling ramps per cycle)步数与加热过程的温度梯度步数相等。
Dynamics steps per ramp:每一温度梯度的动力学步数。
Total number of steps:Annealing cycles ×(Heating ramps per cycle + cooling Heating ramps per cycle)× Dynamics steps per ramp(即上图中的总步数=5×10×500)设置好数据后,点击More…,出现图3-39所示对话框。
图3-39 退火动力学选项图3-40 Advanced选项目标温度根据快冷得到700K的结构而设定为700K,中间最高温度(Mid-cycle temperature)分别设为900K、880K、860K、850K、840K、835K、830K、825K、820K、810K十组数据。。
Advanced选项卡中需要注意的是要选上Include forces。见图3-40。图3-41和3-42分别是在830K和840K温度下退火所得结构。
图3-41 830K退火结构图3-42 840K退火结构
图3-43 830K径向分布函数曲线
单击此处编辑母版标题样式
单击此处编辑母版文本样式 第二级 第三级 第四级 第五级
Materials Studio分子模拟软件
Version 2011
Copyright ?2010, Neotrident Technology Ltd. All rights reserved.
虚拟 “实验”(分子模拟技术)
C R E A T V I T Y
决定依据
单击此处编辑母版标题样式
单击此处编辑母版副标题样式
虚拟设计
表征材料结构,以及与结构相关的性质 —— 解释 设计材料结构,以及与结构相关的性质 —— 预测
Materials Studio是整合的计算模拟平台
? 可兼顾科研和教学需求 ? 可在大规模机群上进行并行计算 ? 客户端-服务器 计算方式 – Windows Linux Windows, – 最大限度的使用已有IT资源 – – – – – – – – DFT及半经验量子力学 线形标度量子力学 分子力学 QM/MM方法 介观模拟 统计方法 分析仪器模拟 …… ? 全面的应用领域 - 固体物理与表面化学 - 催化、分离与化学反应 - 半导体功能材料 - 金属与合金材料 - 特种陶瓷材料 - 高分子与软材料 - 纳米材料 - 材料表征与仪器分析 - 晶体与结晶 - 构效关系研究与配方设计 - ……
单击此处编辑母版标题样式 ? 包含多种计算方法
单击此处编辑母版副标题样式
Materials Studio?
Menu Toolbar
单击此处编辑母版标题样式 Property
View
单击此处编辑母版副标题样式
Job s us status
Project
Materials studio简介 1、诞生背景美国Accelrys公司的前身为四家世界领先的科学软件公司――美国Molecular Simulations Inc.(MSI)公司、Genetics Computer Group(GCG)公司、英国Synopsys Scient ific系统公司以及Oxford Molecular Group(OMG)公司,由这四家软件公司于2001年6月1日合并组建的Accelrys公司,是目前全球范围内唯一能够提供分子模拟、材料设计以及化学信息学和生物信息学全面解决方案和相关服务的软件供应商。 Accelrys材料科学软件产品提供了全面完善的模拟环境,可以帮助研究者构建、显示和分析分子、固体及表面的结构模型,并研究、预测材料的相关性质。Accelrys的软件是高度模块化的集成产品,用户可以自由定制、购买自己的软件系统,以满足研究工作的不同需要。Accelrys软件用于材料科学研究的主要产品包括运行于UNIX工作站系统上的Cerius2软件,以及全新开发的基于PC平台的Materials Studio软件。Accelrys材料科学软件被广泛应用于石化、化工、制药、食品、石油、电子、汽车和航空航天等工业及教育研究部门,在上述领域中具有较大影响的世界各主要跨国公司及著名研究机构几乎都是Accelrys产品的用户。 2、软件概况 Materials Studio是专门为材料科学领域研究者开发的一款可运行在PC上的模拟软件。它可以帮助你解决当今化学、材料工业中的一系列重要问题。支持Windows 98、2000、NT、Unix以及Linux等多种操作平台的Materials Studio使化学及材料科学的研究者们能更方便地建立三维结构模型,并对各种晶体、无定型以及高分子材料的性质及相关过程进行深入的研究。 多种先进算法的综合应用使Materials Studio成为一个强有力的模拟工具。无论构型优化、性质预测和X射线衍射分析,以及复杂的动力学模拟和量子力学计算,我们都可以通过一些简单易学的操作来得到切实可靠的数据。 Materials Studio软件采用灵活的Client-Server结构。其核心模块Visualizer运行于客户端PC,支持的操作系统包括Windows 98、2000、NT;计算模块(如Discover,Amorphous,Equilibria,DMol3,CASTEP等)运行于服务器端,支持的系统包括Windows2000、NT、SGIIRIX以及Red Hat Linux。浮动许可(Floating License)机制允许用户将计算作业提交到网络上的任何一台服务器上,并将结果返回到客户端进行分析,从而最大限度地利用了网络资源。 任何一个研究者,无论是否是计算机方面的专家,都能充分享用Materials Studio软件所带来的先进技术。Materials Studio生成的结构、图表及视频片断等数据可以及时地与其它PC软件共享,方便与其他同事交流,并能使你的讲演和报告更加引人入胜。 Materials Studio软件能使任何研究者达到与世界一流研究部门相一致的材料模拟的能力。模拟的内容包括了催化剂、聚合物、固体及表面、晶体与衍射、化学反应等材料和化学研究领域的主要课题。 3、模块简介 Materials Studio采用了大家非常熟悉的Microsoft标准用户界面,允许用户通过各种控制面板直接对计算参数和计算结果进行设置和分析。目前,Materials Studio软件包括如下功能模块: Materials Visualizer: 提供了搭建分子、晶体及高分子材料结构模型所需要的所有工具,可以操作、观察及分析结构模型,处理图表、表格或文本等形式的数据,并提供软件的基本环境和分析工具以及支持Materials Studio的其他产品。是Materials Studio产品系列的核心模块。
Materials Studio是Accelrys专为材料科学领域开发的可运行于PC机上的新一代材料计算软件,可帮助研究人员解决当今化学及材料工业中的许多重要问题。Materials Studio 软件采用Client/Server结构,客户端可以是Windows 98、2000或NT系统,计算服务器可以是本机的Windows 2000或NT,也可以是网络上的Windows 2000、Windows NT、Linux或UNIX系统。使得任何的材料研究人员可以轻易获得与世界一流研究机构相一致的材料模拟能力。 Materials Studio 由分子模拟软件界的领先者--美国ACCELRYS公司在2000年初推出的新一代的模拟软件Materials Studio,将高质量的材料模拟带入了个人电脑(PC)的时代。 Materials Studio是ACCELRYS 公司专门为材料科学领域研究者所涉及的一款可运行在PC上的模拟软件。他可以帮助你解决当今化学、材料工业中的一系列重要问题。支持Windows98、NT、Unix以及Linux等多种操作平台的Materials Studio使化学及材料科学的研究者们能更方便的建立三维分子模型,深入的分析有机、无机晶体、无定形材料以及聚合物。 任何一个研究者,无论他是否是计算机方面的专家,都能充分享用该软件所使用的高新技术,他所生成的高质量的图片能使你的讲演和报告更引人入胜。同时他还能处理各种不同来源的图形、文本以及数据表格。 多种先进算法的综合运用使Material Studio成为一个强有力的模拟工具。无论是性质预测、聚合物建模还是X射线衍射模拟,我们都可以通过一些简单易学的操作来得到切实可靠的数据。灵活方便的Client-Server结构还是的计算机可以在网络中任何一台装有NT、Linux或Unix操作系统的计算机上进行,从而最大限度的运用了网络资源。 ACCELRYS的软件使任何的研究者都能达到和世界一流工业研究部门相一致的材料模拟的能力。模拟的内容囊括了催化剂、聚合物、固体化学、结晶学、晶粉衍射以及材料特性等材料科学研究领域的主要课题。 Materials Studio采用了大家非常熟悉Microsoft标准用户界面,它允许你通过各种控制面板直接对计算参数和计算结构进行设置和分析。 模块简介: 基本环境 MS.Materials Visualizer 分子力学与分子动力学 MS.DISCOVER https://www.doczj.com/doc/e86917209.html,PASS
1、诞生背景美国Accelrys公司的前身为四家世界领先的科学软件公司――美国Molecular Simulations Inc.(MSI)公司、Genetics Computer Group(GCG)公司、英国Synopsys Scient ific 系统公司以及Oxford Molecular Group(OMG)公司,由这四家软件公司于2001年6月1日合并组建的Accelrys公司,是目前全球范围内唯一能够提供分子模拟、材料设计以及化学信息学和生物信息学全面解决方案和相关服务的软件供应商。 Accelrys材料科学软件产品提供了全面完善的模拟环境,可以帮助研究者构建、显示和分析分子、固体及表面的结构模型,并研究、预测材料的相关性质。Accelrys的软件是高度模块化的集成产品,用户可以自由定制、购买自己的软件系统,以满足研究工作的不同需要。Accelrys软件用于材料科学研究的主要产品包括运行于UNIX工作站系统上的Cerius2软件,以及全新开发的基于PC平台的Materials Studio软件。Accelrys材料科学软件被广泛应用于石化、化工、制药、食品、石油、电子、汽车和航空航天等工业及教育研究部门,在上述领域中具有较大影响的世界各主要跨国公司及著名研究机构几乎都是Accelrys产品的用户。 2、软件概况 Materials Studio是专门为材料科学领域研究者开发的一款可运行在PC上的模拟软件。它可以帮助你解决当今化学、材料工业中的一系列重要问题。支持Windows 98、2000、NT、Unix以及Linux等多种操作平台的Materials Studio使化学及材料科学的研究者们能更方便地建立三维结构模型,并对各种晶体、无定型以及高分子材料的性质及相关过程进行深入的研究。 多种先进算法的综合应用使Materials Studio成为一个强有力的模拟工具。无论构型优化、性质预测和X射线衍射分析,以及复杂的动力学模拟和量子力学计算,我们都可以通过一些简单易学的操作来得到切实可靠的数据。 Materials Studio软件采用灵活的Client-Server结构。其核心模块Visualizer运行于客户端PC,支持的操作系统包括Windows 98、2000、NT;计算模块(如Discover,Amorphous,Equilibria,DMol3,CASTEP等)运行于服务器端,支持的系统包括Windows2000、NT、SGIIRIX以及Red Hat Linux。浮动许可(Floating License)机制允许用户将计算作业提交到网络上的任何一台服务器上,并将结果返回到客户端进行分析,从而最大限度地利用了网络资源。 任何一个研究者,无论是否是计算机方面的专家,都能充分享用Materials Studio软件所带来的先进技术。Materials Studio生成的结构、图表及视频片断等数据可以及时地与其它PC软件共享,方便与其他同事交流,并能使你的讲演和报告更加引人入胜。 Materials Studio软件能使任何研究者达到与世界一流研究部门相一致的材料模拟的能力。模拟的内容包括了催化剂、聚合物、固体及表面、晶体与衍射、化学反应等材料和化学研究领域的主要课题。 3、模块简介 Materials Studio采用了大家非常熟悉的Microsoft标准用户界面,允许用户通过各种控制面板直接对计算参数和计算结果进行设置和分析。目前,Materials Studio软件包括如下功能模块: Materials Visualizer: 提供了搭建分子、晶体及高分子材料结构模型所需要的所有工具,可以操作、观察及分析结构模型,处理图表、表格或文本等形式的数据,并提供软件的基本环境和分析工具以及支持Materials Studio的其他产品。是Materials Studio产品系列的核心模块。
《计算材料学》实验讲义 实验一:Materials Studio软件简介及基本操作 一、前言 1.计算材料学概述 随着科学技术的不断发展,科学研究的体系越来越复杂,理论研究往往不能给出复杂体系解析表达,或者即使能够给出解析表达也常常不能求解,传统的解析推导方法已不敷应用,也就失去了对实验研究的指导意义。反之,失去了理论指导的实验研究,也只能在原有的工作基础上,根据科研人员的经验理解、分析与判断,在各种工艺条件下反复摸索,反复实验,最终造成理论研究和实验研究相互脱节。近年来,随着计算机科学的发展和计算机运算能力的不断提高,为复杂体系的研究提供了新的手段。 在材料学领域,随着对材料性能的要求不断的提高,材料学研究对象的空间尺度在不断变小,纳米结构、原子像已成为材料研究的内容,对功能材料甚至要研究到电子层次,仅仅依靠实验室的实验来进行材料研究已难以满足现代新材料研究和发展的要求。然而计算机模拟技术可以根据有关的基本理论,在计算机虚拟环境下从纳观、微观、介观、宏观尺度对材料进行多层次研究,进而实现材料服役性能的改善和材料设计。因此,计算材料学应运而生,并得到迅速发展,目前已成为与实验室实验具有同样重要地位的研究手段。 计算材料学是材料科学与计算机科学的交叉学科,是一门正在快速发展的新兴学科,是关于材料组成、结构、性能、服役性能的计算机模拟与设计的学科,是材料科学研究里的“计算机实验”。计算材料学主要包括两个方面的内容:一方面是计算模拟,即从实验数据出发,通过建立数学模型及数值计算,模拟实际过程;另一方面是材料的计算机设计,即直接通过理论模型和计算,预测或设计材料结构与性能。计算材料科学是材料研究领域理论研究与实验研究的桥梁,不仅为理论研究提供了新途径,而且使实验研究进入了一个新的阶段。 计算材料学的发展是与计算机科学与技术的迅猛发展密切相关的。从前,即便使用大型计算机也极为困难的一些材料计算,如材料的量子力学计算等,现在使用微机就能够完成,可以预见,将来计算材料学必将有更加迅速的发展。另外,随着计算材料学的不断进步与成熟,材料的计算机模拟与设计已不仅仅是材料物理以及材料计算理论学家的热门研究课题,更将成为一般材料研究人员的一个重要研究工具。由于模型与算法的成熟,通用软件的出现,
铁基块体非晶合金-纳米晶转变的动力学模拟过程 Discover模块 1 原子力场的分配 在使用Discover模块建立基于力场的计算中,涉及几个步骤。主要有:选择力场、指定原子类型、计算或指定电荷、选择non-bond cutoffs。 在这些步骤中,指定原子类型和计算电荷一般是自动执行的。然而,在某些情形下需要手动指定原子类型。原子定型使用预定义的规则对结构中的每个原子指定原子类型。在为特定的系统确定能量和力时,定型原子使工作者能使用正确的力场参数。通常,原子定型由Discover使用定型引擎的基本规则来自动执行,所以不需要手动原子定型。然而,在特殊情形下,人们不得不手动的定型原子,以确保它们被正确地设置。 图 3-1 1)计算并显示原子类型:点击Edit→Atom Selection,如图所示
弹出对话框,如图所示 从右边的…的元素周期表中选择Fe,再点Select,此时所建晶胞中所有Fe原子都将被选中,原子被红色线圈住即表示原子被选中。再编辑集合,点击Edit→Edit Sets,如图所示 弹出对话框见图,点击New...,给原子集合设定一个名字。这里设置为Fe,则3D视图中会显示“Fe”字样,再分配力场: 在工具栏上点击Discover按钮,从下拉列表中选择Setup,显示Discover Setup对话框,选择Typing选项卡。
图3-2 Discover Setup对话框Typing选项卡 在Forcefield types里选择相应原子力场,再点Assign(分配)按钮进行原子力场分配。注意原子力场中的价态要与Properties Project里的原子价态(Formalcharge)一致。 2力场的选择 1)Energy 力场的选择: 力场是经典模拟计算的核心,因为它代表着结构中每种类型的原子与围绕着它的
目 录 Q1:为什么使用Discover进行Dynamics计算时,如果设定了Pressure=1GPa,在计算结果中会出现Pressure等于0,而Stress的XX、YY、ZZ方向为1GPa的情况? (4) Q2:如何在Discover计算中分别对相同环境原子分配不同力场类型? (4) Q3:如何在CASTEP计算中限制某个原子的移动方向? (4) Q4:在安装新的MS时,事先没有停掉License Server,在卸载、安装MS后,发现MS的License Server 无法正常启动。 (5) Q5:如何修改Windows或者Linux下的端口号: (5) Q6:如何使用DMol3进行动力学计算? (6) Q7:如何让Discover程序输出.arc文件? (7) Q8:如何使用rattle关键词来限制水分子的几何结构? (7) Q9,如何使用Standalone方式运行DMol程序? (7) Q10:如何在DMol中加入外界电场? (7) Q12,如何以Standalone方式运行Discover作业? (8) Q13:为什么我在QSAR模块中无法找到新加入的Jurs和DMol3描述符? (8) Q14:如何在DMol模块中,对某一分子只允许其沿着Z方向进行优化,而XY方向则不变? (8) Q15:如果CASTEP计算过程中断电,怎么能够重新开始计算呢?在Keywords中有两个关键词Reuse 和Continuation,它们有什么差异呢? (8) Q16:如果我在Cleave一个平面的时候,选择的是(111)面,或者该晶体原来就是一个三斜晶胞,我怎么才能切出一个长方形的表面来呢? (9) Q17:在使用DMol进行结构优化的时候失败,通过对轨迹的回放发现,整个分子在平面上下进行翻转,并由此导致能量振荡,这种情况应当如何处理? (9)
Materials Studio 5.5软件安装说明 (注:如果之前您的电脑上装有Materials Studio 5.0,请将其卸载后再进行此安装过程) Step 1: 首先将Materials Studio 5.5软件安装程序下载至您的电脑。Materials Studio 5.5软件安装程序具体下载地址:登录东华大学主页(https://www.doczj.com/doc/e86917209.html,/)/材料科学与工程学院主页( https://www.doczj.com/doc/e86917209.html,/)左下角/下载中心/研究生教学课件/ Materials Studio 5.5软件安装程序/下载。 或直接链接:https://www.doczj.com/doc/e86917209.html,/mainAction.do?topNav=159&sideNav=187下载“Materials Studio 5.5软件安装程序” Step 2:打开控制面板/Windows 防火墙/关闭/确定(建议安装前将360等杀毒软件也关闭)。 Step 3:将Materials Studio 5.5软件安装程序解压缩(一般装至非系统盘),点击安装文件setup.exe。
点击Next。 点击Next。
改变安装路径(点击Change),尽量安装至非系统盘,点击Next。 选择Complete,点击Next。
选择Start the Gateway now, with default security settings.选项,点击Next。 选择Install。 Step 4: 打开目录C:\WINDOWS\system32\drivers\etc,用记事本或写字本打开hosts文件,在文本最后一行下面添加219.228.78.3 SERVER,保存,关闭。
Materials Studio6.0软件客户端的安装 1.在每台客户端,先修改文件C:\WINDOWS\system32\drivers\etc\hosts,在其末尾加入一 行:License服务器IP地址 License服务器机器名,如: 192.168.0.1 Server 2.将光盘上的MaterialsStudio60.exe文件复制到硬盘上,比如D:\ 3.双击MaterialsStudio60.exe,会将该文件解压到D:\ MaterialsStudio60 4.进入D:\ MaterialsStudio60,执行setup.exe,如下图:
5.弹出下面界面后,Next到下一步: 6.弹出下面界面后输入用户名和组织,然后Next到下一步:
7.弹出下面界面后请选择安装目录,空间大约需要1.2G,然后Next到下一步: 8.弹出下面界面后一定要选择Complete(默认),然后Next到下一步:
9.弹出下面界面后选择默认值Start the Gateway now,Next到下一步: 10.在下面界面选择Install后,开始安装,请稍等:
11.弹出下面界面后表示客户端软件安装完成,点击 Finish进入到License配置界面:
12.在下面界面选择 Connect to remote license server进入下一步: 13.弹出下面界面后,如下填写: Host name:输入license服务器的机器名; Port:1715(默认); 不要勾选I have redundant license server 然后Next到下一步:
2012级专业课程设计(Ⅱ) 题目:利用material studio软件制作CaTiO3的晶体结 构 姓名:文美乐 班级:应物1203 学号: 指导教师:王会娴
一、课程设计目的、意义 目的: 1. 根据课堂讲授内容,我们做相应的自主练习,便于消化课堂所讲解的内容。 2. 通过多次反复调试程序进而积累相应的经验。 3. 通过完成要求的课题,逐渐培养学生的编程能力,用计算机解决实际问题的能力。 意义: 1. 有助于加深我们对操作软件MS的理解,通过课程设计,我们可以真正理解其应用方向,应用方法。 2. 有利于我们空间思维的锻炼,MS制图能直接有效地训练学生的立体思维、培养分析问题、解决问题能力。即使是一个简单的空间绘图,依然需要学生有条不理的构思。 3. 有利于培养严谨认真的学习态度,在软件应用过程里,如果不够认真或细心,可能就无法得出正确的运行结果。我们反复调试,反复修改的过程,其实也是对我们认真严谨治学的一个锻炼。 应用: 1.MaterialsStudio 在晶体结构教学中的应用 在涉及晶体结构的课程中,学生往往需要掌握晶体的结构和对称性,但单纯板书式教学对学生的理解作用不大,利用MaterialsStudio 的建模功能可以方便的建立各种晶体的三维模型,直观化的展示其结
构和对称性等特点。 2.MS在能带结构计算中的应用 固体中电子能带结构的计算是固体物理学的主要理论问题,晶体电子能带的理论计算方法很多,但对学生的知识结构要求很高,学生学习起来往往感到无从下手。而只有让学生参与实际晶体的能带结构计算,学生对该部分内容的理解才更加深刻。MaterialsStudio中的CASTEP模块可以完成此方面的内容。CASTEP基于总能量的平面波赝势理论,运用原子数目和种类来预测和计算包括晶格参数、分子对称性、结构性质、能带结构、固态密度、电荷密度和波函数、光学性质。由于避免了繁杂的理论推导,一般学生可以很快上手,把重点放到计算结果的分析和讨论上,激发了学生的学习兴趣。 在X射线衍射教学中的应用 X射线衍射内容在固体物理中占有重要地位,通过X射线衍射实验,人们可以对未知晶体进行结构标定.而一般的固体物理教材只介绍X射线衍射的原理,学生对其应用知之甚少。我们利用Mater ialsStudio中的Reflex模块模拟晶体材料的X光、中子以及电子等多 种粉末衍射图谱,确定晶体的结构、解析衍射数据并用于验证计算和实验结果。 4.MS在理论化学计算软件的应用 在众多材料化学理论计算软件中,MS是比较合适一般学生使用。可视化是深奥的材料周期性结构和结果直观表达。MS的操作简单,
一、Materials Studio软件的主要应用领域包括: ? 金属材料研究 ? 无机非金属材料研 ? 纳米材料研究 ? 高分子及其复合材料研究 ? 表界面研究 ? 化学反应研究 ? 含能材料研究 ? 生物、医药研究 ? 在晶体结构、形貌研究中的应用 ? QSAR 的应用 ? Perl 语言的应用 Accelrys(美国)公司是世界领先的计算科学公司,是一系列用于科学数据的挖掘、整合、分析、模建与模拟、管理和提交交互式报告的智能软件的开发者,是目前全球范围内唯一能够提供分子模拟、材料设计、化学信息学和生物信息学全面解决方案和相关服务的软件供应商,所提供的全面解决方案和科技服务满足了当今全球领先的研究和开发机构的要求。 Materials Studio多尺度分子模拟平台是Accelrys公司(美国)在材料设计领域的核心产品。它融合多种模拟方法,整合多达23 个功能模块,实现从电子结构解析到宏观性能预测的全尺度科学研究。在国内拥有近400家用户,分布在石油、化工、环境、能源、制药、电子、食品、航空航天和汽车等工业领域和教育科研部门;相关的研究工作在Nature、Science等各类权威期刊上发表论文过万篇。 Materials Studio分子模拟软件采用了先进的模拟计算思想和方法,如量子力学(QM)、线性标度量子力学(Linear Scaling QM)、分子力学(MM)、分子动力学(MD)、蒙特卡洛(MC)、介观动力学(MesoDyn)和耗散粒子动力学(DPD)、统计方法QSAR(Quantitative Structure - Activity Relationship )等多种先进算法和X射线衍射分析等仪器分析方法;同时产品提供了界面友好的的模拟环境,研究者能方便地建立三维结构模型,并对各种小分子、纳米团簇、晶体、非晶体以及高分子材料的性质及相关过程进行深入的研究,得到切实可靠的数据。 Materials Studio分子模拟软件支持Windows和Linux操作平台,用户可以自由定制、购买自己的模拟方法和模块,以满足特定领域研究需求。 Materials Studio软件使任何研究者都能得到和世界一流研究部门相一致的材料模拟技术。 二、Materials Studio软件与Pipeline Pilot流程处理平台的整合 三、Materials Studio软件的系统要求
欢迎 欢迎使用Materials Studio Materials Studio是一个采用服务器/客户机模式的软件环境,它为你的PC机带来世界最先进的材料模拟和建模技术。 Materials Studio使你能够容易地创建并研究分子模型或材料结构,使用极好的制图能力来显示结果。与其它标准PC软件整合的工具使得容易共享这些数据。 Materials Studio的服务器/客户机结构使得你的Windows NT/2000/XP,Linux和UNIX服务器可以运行复杂的计算,并把结果直接返回你的桌面。 Materials Studio采用材料模拟中领先的十分有效并广泛应用的模拟方法。Accelry’s的多范围的软件结合成一个集量子力学、分子力学、介观模型、分析工具模拟和统计相关为一体容易使用的建模环境。卓越的建立结构和可视化能力和分析、显示科学数据的工具支持了这些技术。 无论是使用高级的运算方法,还是简单地利用Materials Studio增强你的报告或演讲,你都可以感到自己是在用的一个优秀的世界级材料科学与化学计算软件系统。 易用性与灵活性 Materials Studio可以在Windows 98,Me,NT,2000和XP下运行。用户界面符合微软标准,你可以交互控制三维图形模型、通过简单的对话框建立运算任务并分析结果,这一切对Windows用户都很熟悉。 Materials Studio的中心模块是Materials Visualizer。它可以容易地建立和处理图形模型,包括有机无机晶体、高聚物、非晶态材料、表面和层状结构。Materials Visualizer 也管理、显示并分析文本、图形和表格格式的数据,支持与其它字处理、电子表格和演示软件的数据交换。 Materials Studio是一个模块化的环境。每种模块提供不同的结构确定、性质预测或模拟方法。你可以选择符合你要求的模块与Materials Visualizer组成一个无缝的环境。你也可以把Materials Visualizer作为一个单独的建模和分子图形的软件包来运行。 如果你安装了Materials Studio的其它模块,后台运算既可以运行在本机,也可以通过网络运行在远程主机上。这取决于你建立运算时的选择和运算要求。Materials Studio的客户机/服务器模式支持服务器端运行在Windows NT/2000/XP,Linux或UNIX下,使得你可以最大化利用计算资源。 效率和交流 所以的研究人员都可以从Materials Studio强大功能中获益。这份文档的“演示”部分给出了一些简单的分子和材料的模型。这能使你获得对材料的更好的理解并能创建优秀的图形。与其它Windows软件的协同工作使得能容易地拷贝粘贴这些图形到其它文档。结构和性质的数据能容易地从电子表格和数据库中导入导出。Materials Studio帮助你显示和共享数据。Materials Visualizer也可以安装在研究部门、生产部门、