
?
一通信作者,E Gm a i l :y
i n h u i m i n m i n @163.c o m . 案例分析
D O I :10.3969/j
.i s s n .1672G9455.2019.14.0521例颅内窦组织细胞增生症伴巨淋巴结病的案例分析
张金英1,黄银辉2?,陈雅芳1,林智强2,李弥弥1,杨美丽1,林友榆2,蔡若蔚1
1.福建医科大学附属第二医院神经内科,福建泉州362000;2.福建省晋江市医院神经内科,福建晋江362200一一关键词:窦组织细胞增生症伴巨淋巴结病;一中枢神经系统;一病理学;一影像学
中图法分类号:R 741.04文献标志码:C 文章编号:1672G9455(2019)14G2111G02一一窦组织细胞增生症伴巨淋巴结(R o s a i GD o r f m a n )
病是一种良性组织细胞增生性疾病,因大多发生于淋巴结内,又称窦组织细胞增生伴巨淋巴结病,发病率低,且25%~43%发生于淋巴结外,如眼眶二皮肤二呼吸道二软组织二颅内二脊柱等.原发于中枢神经系统的
R o s a i GD o r f m a n 病在该病中所占构成比低于4%,
主要表现为幕上累及硬脑膜的实性肿块,术前多误诊为脑膜瘤,发生于脑实质内较为罕见,易误诊为淋巴瘤二
脑转移瘤[1
].其病因尚不明确,可能与某些病毒感染
有关,如人疱疹病毒6二E B 病毒二H I V 等,但尚未证实[2]
.本研究对1例颅内R o s a i GD o r f m a n 病案例进行了分析,现报道如下.
1一临床资料
一一患者,男,35岁,主诉 头痛伴发作性视物模糊1月余 ,于入院前1月无明显诱因出现头痛,呈持续性
全颅胀痛,程度中等,尚可忍受,伴发作性左眼视物模糊,持续10s 左右可自行缓解,但反复发作,无畏光二怕声,无恶心二呕吐二头晕,无畏冷二发热,无眼眶胀痛二流泪,无肢体无力二麻木,无人事不省二肢体抽搐等,未
重视诊治,症状无好转.发病以来,精神倦怠,睡眠二
食欲欠佳,二便如常,体质量无明显改变.查体:神志清楚,心二肺二腹体征(-)
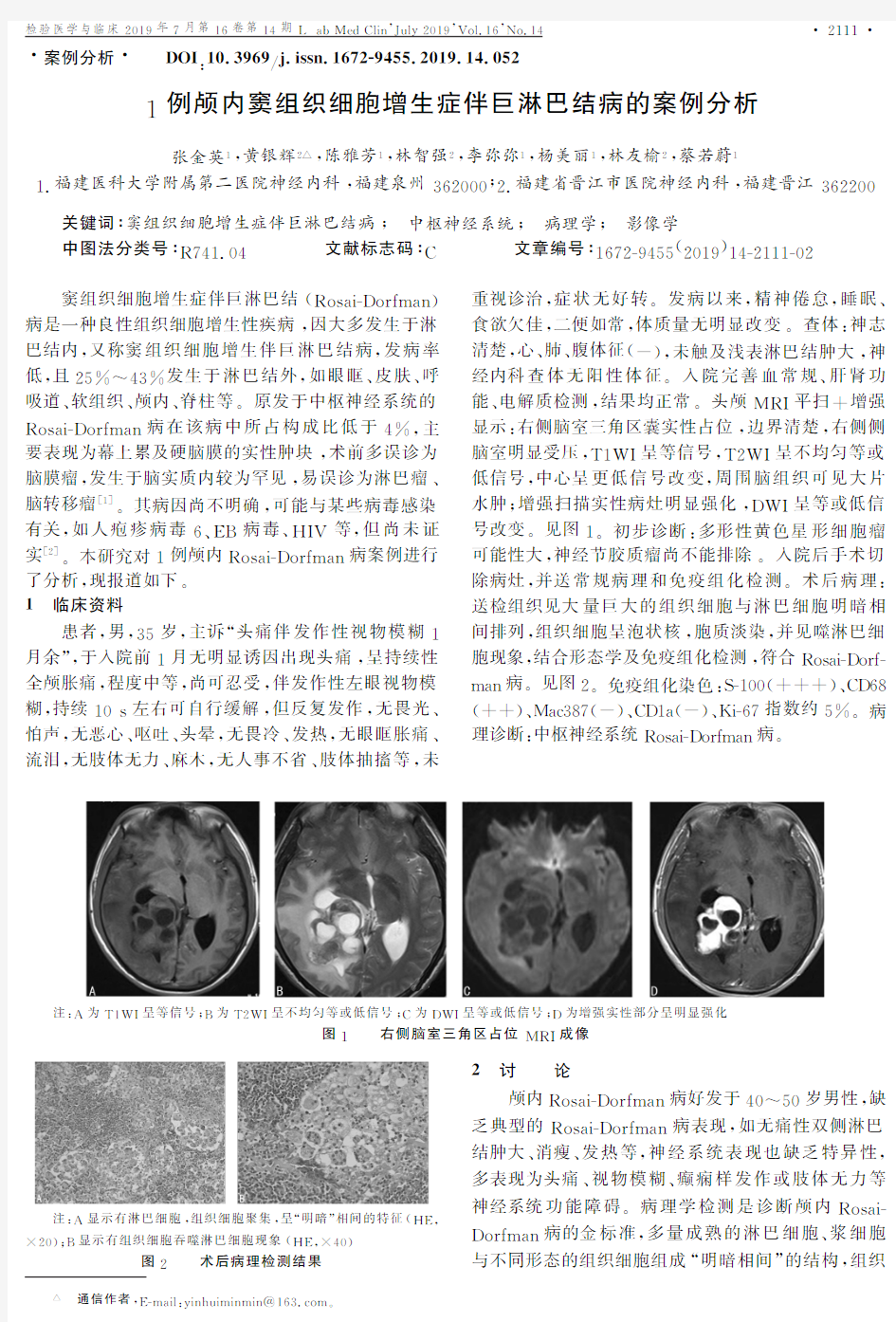
,未触及浅表淋巴结肿大,神经内科查体无阳性体征.入院完善血常规二肝肾功能二电解质检测,结果均正常.头颅MR I 平扫+增强显示:右侧脑室三角区囊实性占位,边界清楚,右侧侧脑室明显受压,T 1W I 呈等信号,T 2W I 呈不均匀等或低信号,中心呈更低信号改变,周围脑组织可见大片水肿;增强扫描实性病灶明显强化,DW I 呈等或低信号改变.见图1.初步诊断:多形性黄色星形细胞瘤
可能性大,神经节胶质瘤尚不能排除.入院后手术切
除病灶,并送常规病理和免疫组化检测.术后病理:送检组织见大量巨大的组织细胞与淋巴细胞明暗相间排列,组织细胞呈泡状核,胞质淡染,并见噬淋巴细胞现象,结合形态学及免疫组化检测,符合R o s a i GD o r f G
m a n 病.见图2.免疫组化染色:S G100(+++)二C D 68(++)二M a c 387(-)二C D 1a (-)二K i G67指数约5%.病
理诊断:中枢神经系统R o s a i GD o r f m a n 病
.
一一注:A 为T 1W I 呈等信号;B 为T 2W I 呈不均匀等或低信号;C 为DW I 呈等或低信号;D 为增强实性部分呈明显强化
图1一一右侧脑室三角区占位M R I
成像
一一注:A 显示有淋巴细胞,组织细胞聚集,呈 明暗 相间的特征(H E ,?20);B 显示有组织细胞吞噬淋巴细胞现象(H E ,?40
)图2一一术后病理检测结果
2一讨一一论
一一颅内R o s a i GD o r f m a n 病好发于40~50岁男性,缺乏典型的R o s a i GD o r f m a n 病表现,如无痛性双侧淋巴结肿大二消瘦二发热等,神经系统表现也缺乏特异性,多表现为头痛二视物模糊二癫痫样发作或肢体无力等
神经系统功能障碍.病理学检测是诊断颅内R o s a i G
D o r f m a n 病的金标准,
多量成熟的淋巴细胞二浆细胞与不同形态的组织细胞组成 明暗相间 的结构,组织
1112 检验医学与临床2019年7月第16卷第14期一L a b M e dC l i n ,J u l y 2
019,V o l .16,N o .14
探讨恶性组织细胞增生症的胸部X线表现 发表时间:2010-06-02T10:54:32.763Z 来源:《中外健康文摘》2010年第4期供稿作者:刘再国 [导读] 恶性组织细胞增生症(malignant histiocytosis,MH)在1939年首先由Scott和Robb-Smith报道 刘再国(黑龙江省伊春市第三人民医院黑龙江伊春 153100) 【中图分类号】R445 【文献标识码】A 【文章编号】1672-5085 (2010)04-0048-02 【摘要】目的探讨恶性组织细胞增生症(MH)的胸部x线表现。方法搜集并整理的17例经骨髓或淋巴结穿刺活检证实的MH患者的胸部X线平片资料。结果胸部x线表现为非间质性病变,渗出性病变,纵隔及淋巴结肿大,肺内肿块,胸腔积液。结论胸部x线表现没有特异性,但是正确认识它对MH的诊断是很有必要的。 【关键词】恶性组织细胞增生症胸部 X线摄影 【Abstract】 Objective of the patient’s condition and symptoms. Malignant histiocytosis Chest RadiographyTodiscuss thoracicX-ray manifestations of malignant histiocytosis. Materials and Methods Plain chest films of 17 patients with pathologically-proved malignant histiocytosis were collected. Radiographic signs were analyzed. Results Radiographic manifestations included interstitial abnormality, exudative shadow, mediastinal and hilar lymphadenopathy,pleural effusion and intra-pulmonary mass.Conclusion Malignant histiocytosis carries no specific radiographic features, evertheless, plain chest filnm is helpful for making a comprehensive understanding 恶性组织细胞增生症(malignant histiocytosis,MH)在1939年首先由Scott和Robb-Smith报道。此病是一种组织细胞及其前体细胞异常增生的恶性疾病。具有全身性、进行性、侵袭性等特点。MH起病急骤,病势凶险,预后极差,患者最后可因全身多脏器衰竭,严重感染,内脏大出血等而死亡。关于MH的x线表现的文献报道并不多。笔者搜集整理并分析本院和其他医院经骨髓或淋巴结穿刺证实的17例MH 患者的胸部平片x线表现,借此来提高对该病x线表现的认识。 1 资料与方法 本组17例,男12例,女5例,年龄4~69岁,平均年龄35岁。其中经骨髓穿刺确诊的15例,经淋巴结穿刺活检确诊的2例。所有患者均有不同程度的发热,以高热较多,且多为不规则热,其中体温最高者达40.9 ℃,体温最低者为37.2 ℃。肝脏肿大15例,脾脏肿大16例,表浅淋巴结肿大5例。咳嗽12例,胸痛8例。有的患者出现腹胀、腹痛、腹水、腹泻、便血、黄疸、肠梗阻等症状。患者入院时血象:白细胞计数低于4.0×109/L者13例,4.0~10.0×109/L者3例,高于10.0×109/L者1例。血红蛋白低于100g/L者15例。血小板计数低于100×109/L者14例。骨髓象:骨髓穿刺涂片找到异常组织细胞(恶性组织细胞)15例,其中部分病例同时见到淋巴样组织细胞、单核样组织细胞、吞噬性组织细胞和少数多核巨组织细胞。淋巴结穿刺:颈部淋巴结穿刺活检2例找到异常组织细胞。 所有患者均摄胸部正侧位片。 2 结果 17例胸部正侧位片15例显示异常,其中7例显示一侧或两侧肺下野云絮状、点状、斑片状密度增高影,边缘较模糊,2例伴有胸腔积液,考虑炎症性改变;4例显示双侧肺中下野网状致密影,其间散在小点状、结节状阴影,考虑肺间质病变;3例显示双侧肺纹理增粗、增强、紊乱;仅1例显示双侧肺门影略大。 3 讨论 3.1 病因 MH病因至今不明,通常认为是组织细胞性淋巴瘤或急性单核细胞性白血病的一种变型,可能与EB 病毒感染有关[1]。亦有人士认为是自身免疫增殖性疾病或者由于免疫功能缺陷所致。近年来报道恶组常作为继发于其他肿瘤的第2个恶性肿瘤。 3.2 病理特点及X线表现 恶性组织细胞广泛浸润是本病病理学的基本特点[1],脾及淋巴结等造血组织为常见,但全身大多数器官组织也可累及,如皮肤、浆膜、肺、心、肾、胰腺、胃肠、内分泌、乳房、睾丸及神经系统等。这些器官及组织不一定每个都被累及,而受累的器官或组织,病变分布亦极不均一。 文献报道恶性组织细胞侵及肺部时,可表现为肺实质和(或)肺间质的浸润性改变[2]。胸片显示肺部云絮状、斑片状边缘模糊的密度增高影,病理上主要是由于肺泡腔内组织细胞浸润,引起肺泡炎;胸片显示肺部网状、结节状阴影,主要是组织细胞在肺泡间隔中明显增生浸润,造成肺泡间隔显著增宽,并伴有淋巴管的扩张;胸片表现球形阴影,是由于组织细胞瘤样增生所致;恶性组织细胞侵及胸膜时,便出现胸腔积液[3]。另外,淋巴结也是其主要侵犯的器官之一。全身各处淋巴结均可受累,但表浅淋巴结较少累及,最常累及的是深部淋巴结,如腹主动脉旁淋巴结、纵隔肺门淋巴结、肠系膜淋巴结等[4]。胸片可显示纵隔增宽和(或)肺门增大[5]。本组所选17例病例的胸部X线表现与文献报道基本一致。 3.3 X线检查在MH诊断中的意义 综合文献资料,笔者认为:在MH诊断中,查找异常组织细胞是确诊的主要依据。胸部X线征象尽管没有特异性,但是紧密结合临床资料综合分析,对本病的诊断也是必要的;MH的临床表现典型时,如果X线检查发现上述征象,尤其是肺部X线征象,而临床又不能用一般感染性疾病来解释者,即使末梢血或者骨髓穿刺检查尚未找到恶性组织细胞,也应想到MH的可能[3]。冀景玲等[6]认为,疑为MH时,作肺部X线检查,对诊断有一定的参考价值。 参考文献 [1]单渊东,张之南.恶性组织细胞病,见:陈敏章主编,中华内科学,北京人民卫生出版社,1999,1:2848. [2]孟繁禄,徐德永等.恶性组织细胞增生症的X线表现(附25例分析),中华放射学杂志,1994,28:627. [3]李邦国,杨志明等.恶性组织细胞增生症的胸部X线表现,临床放射学杂志,2004,23:581. [4]谈顺,吴文川等.恶性组织细胞增生症的临床病理特征.海南医学院学报,1999,5:95. [5]宋兆祺.恶性组织细胞增生症的X线表现(附22例尸检对照)。临床放射学杂志,1982,1:185. [6]冀景玲,李铁一等.成人恶性组织细胞病的肺部X线表现及其病理基础(附4例报告).中华医学杂志,1982,62:407.
组织细胞增生症X(血液系统) 【概述】 儿童时期组织细胞增生症(chilchoodhistiocytosis)是一组以单核-巨噬细胞增生为共同特点的疾病。由于此类疾病的病因与发病机理尚不十分清楚,因而分类比较困难。近年来国际组织细胞协会(HistiocyteSociety)解决了分类和诊断问题,将此类疾病分为三大类(表41-1),即Ⅰ类郎格罕细胞组织细胞增生症;Ⅱ类家族性嗜血性淋巴组织细胞增生症和感染合并嗜血细胞综合征;Ⅲ类恶性组织细胞病和急性单核细胞白血病。对此类疾病不能仅靠临床症状和一般实验室检查,必须作病灶部位的活组织检查方可作出确切的诊断。 表41-1 儿童时期组织细胞增生症分类 分类ⅠⅡⅢ 细胞型郎格罕细胞单核-巨噬细胞 单核-吞噬细胞系的恶性细 胞 细胞功能抗原表达 具有抗原和吞噬细 胞 诊断特异性细胞镜下可见Birbeck颗粒,病灶部位细胞具有OKT6阳 性的单核巨噬细胞特点 组化染色非特异性 脂酶阳性 形态呈恶性组化染色非特 异脂酶阳性 病名LCH 家族性嗜血性网状 细胞增生症恶性网状细胞病、急性单核细胞白血病 一、郎格罕细胞组织细胞增生症
郎格罕细胞组织细胞增生症(Langerhanscell histiocytosis,LCH)以前曾命名为组织细胞增生症x(Histiocytosisx,Hx)多认为是一组反应性增殖性疾病。近年来由于分子生物学、免疫组织化学以及超微结构技术的进展,已将本症的研究工作引向深入,逐渐在分型、诊断、治疗和对预后的评估等方面取得了新的进展。对本症的认识逐渐提高,以北京儿童医院为例,1956~1982年26年中,共收治172例,1982~1991年仅9年中,收治本症178例。 本症是以Langerhans细胞(LC)异常增生为特点。既往根据临床症状将本症分为三种类型,即骨嗜酸肉芽肿,韩薛柯氏综合征和勒雪综合征。骨嗜酸肉芽肿在成人发病多侵犯长骨,而在儿童则多见于颅骨、脊柱、肋骨和骨盆骨,病灶可为单一性或多发性。韩薛柯综合征多见于幼儿和学龄前儿童,以膜性骨的溶骨性改变、突眼和尿崩为常见症状,一般无生命危险,但多呈慢性或病灶进展;勒雪氏病多发生在婴幼儿时期,病情重,以内脏和皮肤、肺和骨骼等多脏器浸润为主,病情进展,病死率较高。由于本症的临床形式表现复杂,预后差别很大,一些病理学家提出是否有一种以上的LCH存在和是否局限形式为"良性"而弥漫性形式为"恶性"等新的研究课题。 本症的发病率尚无确切的统计,据估算发生在一岁内的约为1/10万;15岁以下的约为0.2/10万。此估算一般未包括单一病灶的骨嗜酸肉芽肿和韩雪柯氏综合征。 【病因及发病机制】 病因尚不明确,近年来多认为本症是一种免疫性疾病。自从Basset等首次在本症患者的病变组织中发现Birbeck颗粒,并经多种免疫组织化学方法检查,进一步证实了此种细胞的特异性非常接近正常的LC,由此确立了本症是由于LC异常增生的结果。LC是单核-巨噬细胞系统中的表皮树状突细胞,虽然它的吞噬功能较弱,但在免疫传入系统中起重要作用。它具有T细胞抗原表达和诱发延迟性超敏反应的作用,可分泌具有生物活性的细胞因子,如白细胞介素I(IL-1)和PGE2等,促使破骨细胞功能亢进而发生溶骨现象,造成典型的骨损害症状。
恶性组织细胞病(Malignant Histiocytosis)(恶组)是单核一巨噬细胞系统中组织细胞的恶性增生性疾病。临床表现以发热、肝脾淋巴结肿大、全血细胞减少和进行性衰竭为特征。病理恶性组织细胞浸润是本病病理学的基本特点,脾及淋巴结等造血组织为常见,但全身大多数器官组织也可累及,如皮肤、浆膜、肺、心、肾、胰腺、胃肠、内分泌、乳房、睾丸及神经系统等。这些器官及组织不一定每个都被累及,而受累的器官或组织,病变分布亦极不均一。恶性细胞可以是分散的或成集结,但极少形成瘤样的肿块。被累及的组织中有许多畸形的、形态多样的异常组织细胞,间有多核巨细胞和吞噬性组织细胞,吞噬大量多种血细胞。其异常组织细胞是诊断本病的主要依据。临床表现恶组多见于青壮年,以20~40岁者居多,男女发病为2-3:1.本病按病程可分为急性和慢性型。国内以急性型为多见。起病急骤,病势凶险。发热是最为突出的表现。90%以上病人以发热为首发症状。体温可高达40°C以上。热型以不规则热为多,也有间歇热、弛张热和稽留热。少数病例用抗生素能暂时使体温下降,但更多病例发热与疾病本身有关,对抗生素治疗无反应。皮质激素虽有降温作用,但不持久,只有化疗有效时体温才能恢复正常。贫血也是较常见症状之一。急性型早期即出现贫血,呈进行性加重。晚期病例,面色苍白和全身衰竭非常显著。少数起病缓慢的病例,其最早出现的突出症状可为贫血和乏力。出血以皮肤瘀点或瘀斑为多见。其次为鼻衄、齿龈出血、粘膜血泡、尿血、呕血或便血也可发生。此外,乏力、食欲减退、消瘦、衰弱也随病情进展而显著。肝、脾、淋巴结肿大不一定同时发生。脾大比肝大更为常见。晚期病例,脾肿大可超过脐水平而达下腹。肝肿大一般为轻度到中度,可有压痛,有时被误诊为肝脓肿。淋巴结肿大不如肝脾肿大常见,出现也较晚,以颈、腋下和腹股沟外淋巴结肿大为多见。也可有肠系膜、腹膜后和纵隔等处深部淋巴结受累。不典型病例(特殊类型恶组)可困身体某一组织或器官的病变特别突出,某些特殊的症状或体征成为主要的临床表现,而贫血、出血、脾大等典型表现则不明显。例如有些病例早期主要表现为皮肤结节或肿块;有的出现胸腔积液、腹水、腹痛、腹泻、便血、黄疸、肠梗阻或肠穿孔;有的出现各种神经系统症状如肢体麻木、瘫痪、癫痫等。这些病例可分别称为皮肤型、多浆膜型、胃肠型、神经型恶组等。
纵隔巨大淋巴结增生症1例并相关文献复习 发表时间:2015-07-23T08:58:34.370Z 来源:《医药前沿》2015年第9期供稿作者:高存田辉岳韦名李林李树海司立博 [导读] 巨大淋巴结增生症(Giant Lymph Node Hyperplasia)是由Castleman于1954年首次描述,故又称Castleman综合征[1]。 高存田辉岳韦名李林李树海司立博 (山东大学齐鲁医院山东济南 250012) 【关键词】纵隔巨大淋巴结增生症;诊断与治疗 【中图分类号】R55 【文献标识码】A 【文章编号】2095-1752(2015)09-0351-02 巨大淋巴结增生症(Giant Lymph Node Hyperplasia)是由Castleman于1954年首次描述,故又称Castleman综合征[1]。本病是一种罕见的、原因不明的淋巴结增生性病变,最常发生的部位是胸内纵隔区域,诊断多通过X线及CT检查等,确诊则需要病理组织学。本病治疗首选手术切除。因为本病临床上极少见,且临床表现及影像学表现均没有特异性,在临床工作中常常因为对该病的认识不足而导致误诊误治。本文报告1例纵隔巨大淋巴结增生症患者的临床资料并进行文献复习,以提高对本病的认识和诊疗。 1.临床资料 患者男性,37岁,因“咳嗽1年余,痰中带血2月”入院。行心电图示大致正常心电图;肺功能示:通气功能中度阻塞性减退。血液学检查未见明显异常。行胸部强化CT示:右中后纵隔异常密度阴影,边缘较清楚,密度不均匀。右肺门及纵隔淋巴结轻度肿大。(见图1-2) 图1 图2 经过积极术前准备,患者于入院第7天行右中后纵隔肿瘤切除术,术中见肿瘤位于右中后纵隔,与右支气管稍粘连, 与上腔静脉无粘连,将肿瘤完整切除。术后石蜡病理及酶标示:纵隔巨大淋巴结增生症,肿瘤大小8×6×4cm。CD5(+),Ki-67(+),TDT(-),BCL-2(-),CyclinD1(-),CK(-),CD20(+)。 术后患者恢复良好,顺利出院。 2.讨论 2.1 病因和发病机制 巨大淋巴结增生症的病因及发病机制目前尚不明确。有研究证实,在巨大淋巴结增生症中存在人疱疹病毒-8(HHV - 8) 序列, 提示HHV -8 在其病理过程中可能起一定作用[3]。而近年来学者们则更注重于对HHV28 感染和IL- 26过度表达的研究[4]。 巨大淋巴结增生症可发生于全身任何部位,其发生率由高到低依次是胸部、颈部、腹部、腋部。绝大多数的胸部巨大淋巴结增生症发生于纵隔,最常见于中纵隔和肺门,其次是前纵隔和后纵隔[5]。Keller曾报道了81例经临床病理确定的巨大淋巴结增生症,其中绝大多数是发生于纵隔内[6]。本病多见于青中年患者,发病的高峰期为30~40岁[7],没有明显性别差异。 2.2 临床表现 巨大淋巴结增生症按病变的范围可分为两型:局限型和弥漫型。局限型临床上多无症状,多为体检时发现。弥漫型通常有临床症状与体征,如发热、贫血表现、淋巴结肿大、肝脾肿大、血沉加快等。 本病的诊断多通过X线、胸部CT及MRI。 1).X线:病变好发于纵隔和肺门,常表现为纵隔增宽,边缘光滑、密度均匀。病变多数为单发,多发者肺门也可见肿块。 2).胸部CT:平扫表现为肺门纵隔旁圆形、类圆形或分叶状软组织肿块密度影。增强扫描示病变强化明显,外周可有小点样异常增强的血管影,这与病灶血管增生与毛细血管异常增生、扭曲和扩展有关[8]。 3).MRI: T1WI为低于骨骼的低信号灶,T2WI为显著的高信号影, Gd-DTPA静脉注射后多数肿块出现中等至明显的不均匀强化,其中还可见低信号的纤维间隔[9]。 2.3 治疗与预后 纵隔巨大淋巴结增生症只要诊断明确就可手术。手术彻底切除后可获治愈,但如果首次手术切除不彻底就有复发的可能,复发者也可再次手术治疗。本病预后良好,术后很少复发,有文献报道本病5 年生存率可达100%。 【参考文献】 [1] Castleman G, Iverson L, Menendez V: Localized mediastinal lymph node hyperplasia resembling thymoma. Cancer 9 : 822-830, 1956. [2]Seo HY, Kim EB, Kim JW, et al. Complete remission in a patient with human herpes virus-8 negative multicentric Castleman
恶性组织细胞病的诊断标准 *导读:对不明原因的长期发热而不能以感染性疾病解释者,尤其是伴有全血细胞减少和肝、脾、淋巴结肿大时,应考虑本病的可能性;结合血象、骨髓象或淋巴结活检中找到大量异形或多核巨组织细胞,可以确立诊断。…… 本病的临床表现多样化,缺乏特异性,应密切结合实验室检查综合诊断。实验室检查以骨髓涂片发现异形组织细胞或多核巨组织细胞最为重要,单纯发现吞噬性组织细胞增多不能确定诊断。由于病变呈局灶性,必须反复多部位骨髓穿刺。有报告胸骨穿刺阳性率较高。外周血片异常组织细胞检出率不高,但血液离心后的白细胞层涂片观察可提高阳性率。淋巴结的病理学改变显著,浅表淋巴结活检又较方便,但必要时仍须多部位病理检查,以免漏诊。 应注意与感染性疾病所致的反应性组织细胞增多症相鉴别。反应性组织细胞增生呈良性过程,骨髓中所见的组织细胞多为正常形态,大小较为一致;在原发病如伤寒、粟粒性结核、病毒性肝炎及症疾等治愈后,组织细胞增生将会消退,且中性粒细胞碱性磷酸酶活性大多正常或升高。噬血细胞性组织细胞增多症的骨髓中可见到吞噬红细胞、粒细胞或血小板的组织细胞,但噬血活性不是恶性组织细胞的特点。淋巴瘤特别是 CD30 + 的间变性大细胞淋巴瘤 (Ki-1 性的 T 细胞淋巴瘤 ) 与恶组在临床上、组织
病理上易发生混淆,此时免疫组化染色 CD68 + 、 CD30 - ,且无 T 细胞、 B 细胞表型,有助于确定异常细胞的组织细胞来源。 1 、血象大多呈全血细胞减少。早期即有贫血,多为中度。半数以上白细胞4.0×109/L ,可低达1.0×109/L。少数患者早期白细胞可增多。疾病早期可有中至重度贫血,血小板减少。随着疾病的进展,多数有进行性全血细胞减少。仅少数患者(17.71%) 周围血片中可见少量异常组织细胞,因为组织细胞在造血组织中被网状纤维所固定,细胞间相互粘附,不易进入周围血液。 2 、骨髓检查多数增生活跃,增生低下病例多已达晚期。多数病例骨髓中能找到多少不一样的异常组织细胞。多数增生活跃,增生差者表示病情已严重。多数病例骨髓中找到数量不等散在或成堆的异常组织细胞。异常组织细胞的分类尚不统一,一般分为以下三型:①异形组织细胞:细胞体积较大,形态奇特;胞浆比一般原始细胞丰富,蓝色,可有伪足,并有空泡;核不规则,有时呈分叶状,偶有双核,核仁隐显不一,有时较大。②多核巨组织细胞:大小似巨核细胞,外形不规则,通常含 3~6 个核,彼此贴近或呈分叶状,核仁清晰。③吞噬性组织细胞:其形态与正常巨噬细胞类同,浆内常吞噬大量血细胞,包括幼红细胞、成熟红细胞碎片、血小板,偶有少数中幼粒细胞。异形组织细胞和(或)多核巨组织细胞对恶组有诊断意义。吞噬性组织细胞在其他疾病也可出现,因此缺乏特异性诊断价值。 异常组织细胞酸性磷酸酶阳性,可被酒石酸所抑制。非特异性酶
巨淋巴结增生症的影像表现与分析 【摘要】目的:提高巨淋巴结增生症(GLNH)的认识水平,探讨GLNH 的CT表现及诊断价值。方法:通过1例经手术及病理证实的GLNH的病例分析,结合文献复习,对本病的影像诊断与鉴别诊断进行讨论。结果:GLNH是一种异源性的罕见的淋巴组织增生性良性疾病,其组织形态上可分为透明血管型和浆细胞型两类,前者相对多见,常表现为纵隔病变,后者好发于肠系膜和腹膜后淋巴结。增强后有不同程度增强, 透明血管型呈中等或明显强化,浆细胞型为轻度强化。结论:GLNH具有特征性CT表现,CT具有较高诊断价值,在动脉期明显强化,延迟期持续强化,提示Castleman病的诊断。 【关键词】巨淋巴结增生;影像学;X线;CT;MRI 巨淋巴结增生症(GLNH)是一种少见病,病因尚不明确,患者多无症状,为异源性的罕见的淋巴组织增生性疾病,亦称为血管滤泡性淋巴组织增生。GLNH 于1954年由Castleman首先发现,故又称其为Castleman病。现报告1例经手术及病理证实的GLNH,结合文献资料[1,2]对其病因、临床表现、病理、诊断与鉴别诊断、治疗及预后进行讨论。 1 病例报告 患者马某,男,48岁。因颈部肿块1个月入院,无明显不适。CT示:左上颈部胸锁乳突肌内缘见一类圆形软组织肿块影,大小约3 cm×4 cm,边缘光滑,增强后明显强化,CT值约158 Hu,肿块内见无强化区,病灶与周围结构分界清晰。病理诊断:GLNH。 2 讨论 2.1 发病与病因 GLNH自1954年发现以来,截至1991年的资料,共有230余例报道[3,4]。关于GLNH的病因尚存在两种不同的学说。第一种认为本病表现为反应性淋巴组织增生,与慢性抗体刺激有关,可能为病毒感染所致。第二种理论则认为透明血管型是一种淋巴组织的进行性生长紊乱,而浆细胞型的发生与IL?6调节障碍有关。 2.2 临床表现 GLNH中透明血管型最为常见,约占发病率的76%~91%,而浆细胞型则较少见。GLNH按其临床、影像学表现及其预后亦分为2型:局限型和弥漫型。局限型表现为孤立淋巴结或纵隔内某一组淋巴结受累。弥漫型表现为一组以上的淋巴结受累或影像学发现胸外淋巴结受累,此型可累及肺部。局限型临床多无症状,为体检或常规胸片偶尔发现,可见于任何年龄,发病高峰为30岁~40岁之间,女性约4倍于男性,95%以上为透明血管型,手术多可切除,预后良好。弥漫型通常有临床症状与体征,包括发热、乏力、贫血、表浅淋巴结肿大、肝脾肿大、血沉加快、多克隆性高免疫球蛋白血症和骨髓浆细胞病等,可见于任何年龄,发病高峰为40岁~50岁之间,女性约2倍于男性,绝大多数为浆细胞型,手术切除困难,以放疗及激素疗法为主,预后较差。 2.3 影像表现 2.3.1 局限型GLNH的影像学表现 GLNH可发生于全身各部位的淋巴结,沿全身淋巴链分布,但最好发于纵隔、肺门和腹膜后区域。约60%~70%发生于胸部,沿胸内淋巴链分布,最常见于中纵隔和肺门,其次为前纵隔和后纵隔。约20%发生于腹部,以腹膜后最常见。病灶的大小差异较大,X线胸片可显示肿块的部位和形态,但对明确诊断缺乏特异性,常表现为单发边缘清楚的球形或分叶状肿块,类似于胸腺瘤、淋巴瘤和神经源性肿瘤的表现,位于中纵隔和肺门者可致邻
恶性组织细胞病诊断依据及与反应性组织细胞增多症的鉴别 恶性组织细胞病的诊断应有以下依据: 临床长期不规则发热,以高热为主,伴有进行性全身衰竭,肝脾和淋巴结进行性肿大,可伴有黄疸、出血、皮肤损害和浆膜腔积液等。 进行性全血细胞减少,个别患者外周血中有异常组织细胞,或不典型的单核细胞。 骨髓涂片发现多种形态的异常组织细胞。这是本病诊断的主要依据。 异常的组织细胞有以下几种类型: (1) 恶性组织细胞:细胞体积较大、直径34~54μm,可呈圆形、椭圆形、不规则形或狭长而有伪足。胞浆比一般原始细胞丰富,呈蓝色或深蓝色,不含颗粒,较成熟者胞浆为浅蓝色,含有少数嗜天青颗粒,可伴有空泡;核呈圆形、椭圆形、肾形或不规则形,可有分支或切迹,核边缘可呈裙边样,偶有双核;染色质致密呈网状;核仁1~3个不等,大而清晰;有丝分裂多见。 (2) 多核巨组织细胞:胞体大,直径约为50~90μm,外形不规则,胞浆蓝或灰蓝,无颗粒或含有少数细小颗粒;含有多个(2~10)或多叶核,核仁明显,核间有时可见细丝相连。(3) 吞噬组织细胞:形态与一般的组织细胞相似,体积大,外形不规期,单核或多核,椭圆形,偏位,染色质疏松;核仁隐约约可见;胞浆丰富,含有被吞噬的红细胞或其残余碎片、幼红细胞、血小板或中性粒细胞。 除上述几种主要的细胞外,正常形态的组织细胞数量常常也增多;此外,还有一些单核样、淋巴样和浆细胞样组织细胞。 骨髓或肝脾、淋巴结及其它受侵犯组织的病理切片中可见异常组织细胞浸润,这些细胞呈多样性、成灶性或片状,松散分布,组织结构可部分或全部破坏。 临床表现与异常组织细胞的形态特点诊断时同样重要,两者都不能偏废。若临床典型,又有形态学支持就可确诊。如临床支持,形态学欠缺,应该做多部位骨髓穿刺,争取得到形态学的证实。形态学上恶性组织细胞及多核巨细胞为主要诊断依据。能排除反应性组织细胞增多症可诊断此病。
1.什么是恶性组织细胞病?主要特点是什么? 恶性组织细胞病(malignant histocytosis,MH)是组织细胞及其前身细胞异常增生的恶性疾病。 主要特点是:肝、脾、淋巴结、骨髓等器官组织中出现广泛的恶性组织灶性增生,常伴有明显的血细胞被吞噬现象。 2.恶性组织细胞病(恶组)的诊断标准? 1996年,Rappaport提出诊断标准。①无肿瘤形成,而有骨髓、淋巴结、肝、脾等全身造血组织的弥漫性,异常组织细胞增生;②成熟组织细胞不仅有成熟型,也有未成熟型和血细胞吞噬现象。 3.为什么说过去诊断的恶组病例不是真正意义上的恶组? 近10年来随着免疫组化,细胞分子遗传学、分子生物学等现代科技研究的不断开拓,对恶组的诊断有了新的概念。过去诊断为恶组的病例大多数属于T淋巴细胞间变大细胞淋巴瘤,部分为噬组细胞综合征,而真正来源于单核吞噬细胞系统的恶组病例极罕见。 4.恶组作为第二肿瘤其发生原因是什么? 恶组病因至今不明,通常认为是组织细胞淋巴瘤或急性单核细胞白血病的一种变异型。近年来报道恶组作为第二恶性肿瘤。常并发于恶性淋巴瘤。(B细胞型)T细胞和裸细胞性急性淋巴细胞白血病,急性粒单核细胞性白血病,Lennerts淋巴瘤。其原因与化疗和原发性肿瘤免疫抑制,导致染色体异常,克隆恶性突变有关。 5.恶组的血象特征? 典型的恶组病例血象表现为全血细胞进行性下降分类中示中性粒细胞比例降低,偶见幼粒细胞、幼红细胞,少数病例在涂片尾部发现异常组织细胞。浓缩外周血的细胞层,涂片中恶组检出率可高达60%以上。中性粒细胞碱性磷酸酶(NAP)染色的阳性率和积分值明显减低,甚至完全阴性。 6.恶组的髓象特征? 骨髓大多增生活跃,由于恶组细胞浸润常呈局灶性,不均一性,因此多部位穿刺可提高阳性率。主要特征是见到数量不等,形态异常的恶组细胞。 7.恶组细胞的形态学特征可为几型? 恶组病理学及其形态学的共同特征为:多行性、异行性及吞噬性。按其形态为:①恶性组织细胞,特点:各细胞大小、外形、核形及成熟程度等常常很不一致,畸形明显。 细胞畸形程度随着病情进展愈益明显。②多核巨组织细胞,特点:体积特别巨大③巨噬血细胞:特点:含有种类不同,多少不一的被吞噬的血细胞,包括红细胞碎片,有核红细胞和少量中性粒细胞④淋巴样组织细胞⑤单核样组织细胞。 8.细胞形态学诊断恶组的主要依据是什么? 异常样组织细胞及多核巨组织细胞的发现是恶组诊断的重要依据? 9.恶组的细胞化学染色怎样? 恶组细胞过氧化物酶(POX)染色、苏丹黑、碱性磷酸酶(NAP)染色和β葡萄糖醛酸酯酶均为阳性反应。酸性磷酸酶(ALP)染色为中度或强阳性反应,可被酒石酸所抑制。过碘酸-席夫(PAS)染色可为阴性或阳性反应酸酸AS-D荼酚酯酶(NAS-DCE)染色为阳性。β-葡萄糖醛酸酯酶染色多为阳性反应。血清溶酯酶明显升高。免疫组织化学染色可见恶组细胞及入链染色均呈阳性反应。 10.恶组细胞的标志酶是什么? -抗胰(摩)蛋白酶和血管紧张素转换酶为恶组细胞的标志酶。 11.为什么恶组患者血清铁蛋白升高?
121.X 连锁淋巴增生症 概述 X 连锁淋巴增生症(X-linked lymphoproliferative disease,XLP),又称X 连锁淋巴组织增生综合征,包括XLP1 和XLP2 两个亚型。XLP1 是由编码信号转导淋巴细胞活化分子(signaling lymphocyte activation molecule,SLAM)相关蛋白(SLAM-Associated Protein,SAP)的SH2D1A 基因突变所导致的一种X 连锁隐性遗传病,以EBV 感染后的暴发性传染性单核细胞增多症、异常免疫球蛋白血症及B 细胞淋巴瘤为主要临床特点。 XLP2 即X 连锁凋亡抑制因子(X-linked inhibitor of apoptosis,XIAP)缺陷。因其主要临床表现为噬血细胞综合征,部分患者可出现肠道炎症,如克罗恩病或者结肠炎等,但很少发展为淋巴瘤,现多将其称为X 连锁家族性噬血细胞综合征。所以,本文主要对SH2D1A 基因突变导致的XLP1 展开论述。 病因和流行病学 SH2D1A 基因半合子(男性)或纯合突变(女性),导致SAP 蛋白表达减少甚至缺如,不能正常参与免疫细胞间的信号转导,而致T/B 细胞相互作用异常、使NK 细胞介导的细胞毒作用缺陷,最终导致XLP1 的发生。SH2D1A 基因突变类型包括插入、缺失、错义及剪切突变等。值得注意的是,突变类型与临床表型的严重程度不相关。XIAP(又称BIRC4)基因半合子(男性)或纯合突变(女性),导致XIAP 蛋白表达减少甚至缺如。XIAP 蛋白是重要的凋亡抑制因子,但它的减少或缺乏导致XLP2 发生的原因目前仍未知。 XLP1 多见于男童或男性,患病率为1/1000 000~3/1000 000 男性。杂合突变携带的女性多无临床表现,因为X 染色体的随机失活(莱昂化)通常保留下正常的那条X 染色体。而当女性莱昂化不恰当地失活了正常的那条X 染色体,或仅有一条X 染色体(如特纳综合征)时,SH2D1A 杂合突变携带的女性亦可出现XLP1 的临床表现。与XLP1 类似,XLP2 亦多见于男性,患病率约为1/5000 000男性。
腹膜后巨淋巴结增生症的CT表现 发表时间:2018-03-16T14:20:30.717Z 来源:《医师在线》2017年12月下第24期作者:陈丽仙 [导读] 临床上以深部或浅表淋巴结显著肿大为特点,部分病例可伴全身症状和(或)多系统损害,多数病例手术切除肿大的淋巴结后,效果良好[2]。 (曲靖市第一人民医院医学影像科;云南曲靖655000) 摘要目的:研究探讨腹膜后巨淋巴结增生症的CT表现。方法:选择2017年10月本院腹膜后巨淋巴结增生症的患者1例进行研究讨论,对其临床资料以及CT图像进行回顾性分析。结果:患者顺利完成手术,肿物全部切除,病理检查结果显示为巨大淋巴结增生症,透明血管型。结论:对于膜后巨淋巴结增生症的患者,必须要根据病理组织活检的结果来进行诊断,具有特征性的CT增强表现,确诊后进行手术切除,透明血管型CT增强可以确定诊断结果。 关键词:腹膜;巨淋巴结增生症;CT表现 Castleman病(CD)属原因未明的反应性淋巴结病之一,临床较为少见[1],病理特征为明显的淋巴滤泡、血管及浆细胞呈不同程度的增生,临床上以深部或浅表淋巴结显著肿大为特点,部分病例可伴全身症状和(或)多系统损害,多数病例手术切除肿大的淋巴结后,效果良好[2]。CD的病因未明,本次实验选择2017年10月本院腹膜后巨淋巴结增生症的患者1例进行研究讨论,以便于为今后该病的诊断提供有价值的参考依据,具体如下。 1资料与方法 1.1一般资料 患者,女,50岁,因“左侧腹部疼痛半年”就诊。实验室检查:CA724:18.05U/mlH(0-6.9)余肿瘤标记物(-)彩超:左肾右后方脊柱旁探及实质低回声包块,大小约54mmX32mm,似有包膜;CDFI:其内可见多个粗大血流信号;PW引出动静脉频谱,PS:38.4cm/s,RI:0.26。手术治疗,游离输尿管及左侧肾脏下极,充分游离包块后,以结扎钉、止血夹夹闭并离断包块滋养动脉、静脉,完整切除腹膜后包块,再次探查包块周围组织,见腰大肌前方一2×1cm大小肿大淋巴结,游离后切除,置入标本袋,将包块及肿大淋巴结置入标本袋,取出标本送检。 1.2检查方法 使用单层螺旋CT进行扫描,横断位,电压120kv,电流100-150mAs,层距和层厚5mm,指导患者仰卧位,屏气,两手抱头,用高压注射器,注入对比剂碘海醇1.5ml/kg,速度3.0ml/s,完成全腹动脉和门静脉的扫描,由经验丰富的阅片医生至少两名进行分析探讨,记录大小、形态以及位置等。 2结果 患者顺利完成手术,肿物全部切除,病理检查结果显示为巨大淋巴结增生症,透明血管型。CT:腹膜后左肾内侧见一肾形软组织肿块,边缘清楚,大小约59mmx30mm,增强后皮质期明显强化,髓质期及延迟期持续性强化,供血动脉来自椎旁小动脉,引流静脉汇入左肾静脉,左肾受推移向外侧移位,病灶与左肾、肾上腺分界清楚。 3讨论 3.1局灶型 表现为孤立淋巴结或纵隔内某一组淋巴结受累。青年人多见,发病的中位年龄为20岁(男女比例1:4),90%病理上为透明血管型[3]。患者呈单个淋巴结无痛性肿大,生长缓慢,形成巨大肿块,直径自数厘米至20cm左右,可发生于任何部位的淋巴组织。儿童胸部少见,一般好发于颈部、腹部。 3.2多中心型 较少见,表现为一组以上淋巴结受累或影像学发现胸外淋巴结受累。发病年龄靠后,中位年龄为57岁(男女比例1:2)。患者有多部位淋巴结肿大,易波及浅表淋巴结。伴全身症状如发热、乏力贫血、表浅淋巴结肿大及肝脾肿大;常有多系统受累的表现,如肾病综合征、淀粉样变、重症肌无力、周围神经病,颞动脉炎、舍格伦综合征(干燥综合征)、血栓性血小板减少性紫癜等[4]。20%~30%的患者在病程中可并发卡波西肉瘤或B细胞淋巴瘤。多中心型临床常呈侵袭性病程,易伴发感染。 3.3透明血管型 占80%~90%。淋巴结直径为3~7cm,大者可达25cm,显微镜见淋巴结内许多增大的淋巴滤泡样结构,呈散在分布。有数根小血管穿入滤泡,后期小血管呈玻璃样改变,血液在微循环的时间停留延长,延迟扫描强化更明显。 3.4浆细胞型 占10%~20%。患者常伴有全身症状,如发热、乏力、体重减轻、贫血、红细胞沉降率升高、血液丙种球蛋白增高和低白蛋白血症。淋巴结切除后症状可消失。病理呈滤泡性增生但小血管穿入及滤泡周围的淋巴细胞增生远不及透明血管型明显。动脉早期强化不明显,较晚时相呈中度强化。 3.5混合型 兼有上述二型的特点。CT表现:①病灶多为类圆形结节或肿块影;位于颈部间隙、中纵隔的病灶可因生长间隙受限而呈长椭圆形;位于腹腔和后纵隔的病灶往往较大。②病灶于CT平扫多表现为密度均匀,较少发生囊变、坏死。这与CD血供丰富以及淋巴滤泡组织不易坏死的特性有关。③增强后于动脉期明显强化,静脉期或延迟期持续强化;病灶周边可见明显迂曲的供血和引流血管。④部分病灶内可见点状或细条样钙化高密度影,反映了病灶内血管壁的变并钙质沉积。⑤病灶的周边可见“卫星”结节。⑥混合型主要表现为多组淋巴结肿大,密度均匀,增强扫描多表现为轻度或中等程度强化,这与以大滤泡和滤泡间浆细胞浸润为主,而血管增生较少有关。 3.6MR表现 磁共振检查T1WI呈低信号,T2WI呈高信号,质地均匀,坏死少见,增强特征与CT相似。虽然对病灶内部的钙化显示率低,但其高软组织分辨力的特点以及采用动态增强技术,对病变的定位和显示毗邻关系较CT优越,并能清晰显示肿块增强特点。嗜铬细胞瘤:肾上腺区肿块,较大者密度不均,可有囊变坏死及钙化灶,增强后实质部分明显强化。临床表现有阵发性或持续性高血压及代谢紊乱、血、尿中VMA升高。肾上腺淋巴瘤:肾上腺本身并无淋巴组织,受侵肾上腺多为继发、属血行转移的晚期病变。表现为单侧或双侧肾上腺区的肿
淋巴结肿大 淋巴结病变可分为: 非肿瘤性淋巴结肿大(即各种感染和非感染因素产生的反应性增生); 肿瘤性淋巴结肿大; 介于良恶性之间的淋巴结肿大(如Castleman’s病) 一、非肿瘤性淋巴结病变:可分为 1、感染性疾病引起的局部和全身淋巴结病变 (1)局灶性淋巴结病变: ⑴病毒所致:传染性单核细胞增多症,咽结膜炎,流行性角膜炎,巨细胞病毒, 风疹,水痘,带状疱疹,接种后淋巴结炎,生殖期疱疹感染,人类疱 疹病毒-6 ⑵细菌所致:猩红热,化脓,结核,梅毒,猫爪病,软下疳,鼠疫,兔热病,鼠 咬热,炭疽,类鼻疽,鼻疽 ⑶衣原体所致:性病淋病肉芽肿,沙眼 ⑷立克次体所致:斑疹伤寒,立克次体病 ⑸原虫所致:弓形体病,利什曼病,非洲锥形虫病,南美锥形虫病 ⑹肠虫所致:罗阿丝虫病 ⑺真菌所致:组织胞浆菌病,球袍子菌病,副球袍子菌病 (2)广泛性淋巴结病变 ⑴病毒所致:传染性单核细胞增多症,巨细胞病毒,麻疹,传染性肝炎,获得性 免疫缺陷病,猩红热,粟粒性结核,布鲁杆菌病,钩端螺旋体病,梅毒(二期),类鼻疽,鼻疽,沙门氏菌病,麻风 ⑵衣原体所致:性病淋巴肉芽肿 ⑶立克次体所致:斑疹伤寒 ⑷原虫所致:弓形体病,利什曼病,非洲锥形虫病,南美锥形虫病 ⑸真菌所致:组织胞浆菌病,球袍子菌病,副球袍子菌病 2、非感染性淋巴结病变和病因不明的淋巴结肿大: ⑴药物性淋巴结病变(苯妥英纳、卡马西平、头孢菌素、扑米酮、舒林酸、乙胺嘧啶、开博通、水杨酸偶氮磺胺吡啶、阿替洛尔、奎尼丁、金制剂) ⑵硅性淋巴结病变 ⑶Kawasaki 综合症(皮肤粘膜性淋巴结病变,川畸病):本病于1967年由日本
川畸富作首先报告,又称急性发热性皮肤粘膜淋巴结综合症。其主要临床表现是全身血管炎、急性发热和皮疹。80%发生于4岁以下的孩子,1岁左右发病最多。 本病之病因目前尚不大清楚,可能与感染和免疫因素有关,最近认为逆转录病毒可能与本病有关。 临床症状: ①持续5天以上的发热,热型不规则,可达39℃以上,抗生素治疗无效。 ②手足肿硬,手掌和足底潮红。恢复期手指与足趾端脱皮。 ③多形红斑样皮疹,无疱疹及结痂。 ④双眼球结膜炎。 ⑤口唇红、皲裂,草莓样舌,口咽部潮红。 ⑥颈部淋巴结肿大。 川畸病大部分预后良好,问题在于其心血管系统广泛受累及其严重后果。少数病例在急性期可以发生猝死,或遗留冠状动脉病变直至成年。 皮肤性淋巴结病变系慢性瘙痒性皮炎致慢性淋巴结炎的特殊类型,归属于RNSL。肿大淋巴结避免按压或反复触摸,患者应定期随访。 ⑷Kikuchi坏死性淋巴结病变(菊池病、组织细胞坏死性淋巴结炎、亚急性坏死性淋巴结炎、缺乏粒细胞浸润的坏死性淋巴结炎): 首先由日本学者藤本吉秀和菊池昌弘于70年代初报道,80年代初逐渐被认识的一种病因不明(可能与病毒感染诱发的局部高敏反应和自身免疫有关)的非肿瘤性淋巴结肿大,是一种自限性疾病,病情在1~5个月内缓解,多发于年轻人(10-30岁),女性多见。预后良好,少数可复发。 临床三大特征:顽固性发热;淋巴结肿大;一过性白细胞减少。 病前多有上呼吸道感染等表现。临床上可分为单纯型和变态反应型。前者症状典型,病程短,多于3个月内自愈。后者为变应性发热,除了淋巴结外受累脏器较多,可多次复发,病程较长。淋巴结活检可确诊本病抗生素治疗无效,激素治疗效果非常好。部分患者EBV和小肠结肠炎耶尔森菌血清学检查阳性。 ⑸Kimura病,又称为嗜酸性粒细胞增生性淋巴肉芽肿。是东方人反应性淋巴结病变的重要类型。淋巴结肿大主要位于头部、颈部。组织学改变有滤泡中心细胞增生、生发中心血管形成、出现Warthin-Finkeldey型多核细胞、副皮质区毛细血管后微静脉增多、嗜酸粒细胞浸润、硬化。 ⑹组织细胞增多症X(郎格罕细胞组织细胞增生症)