
酵母双杂基础知识总结
酵母双杂原理:酵母双杂交系统由Fields和Song等首先在研究真核基因转录调控中建立。典型
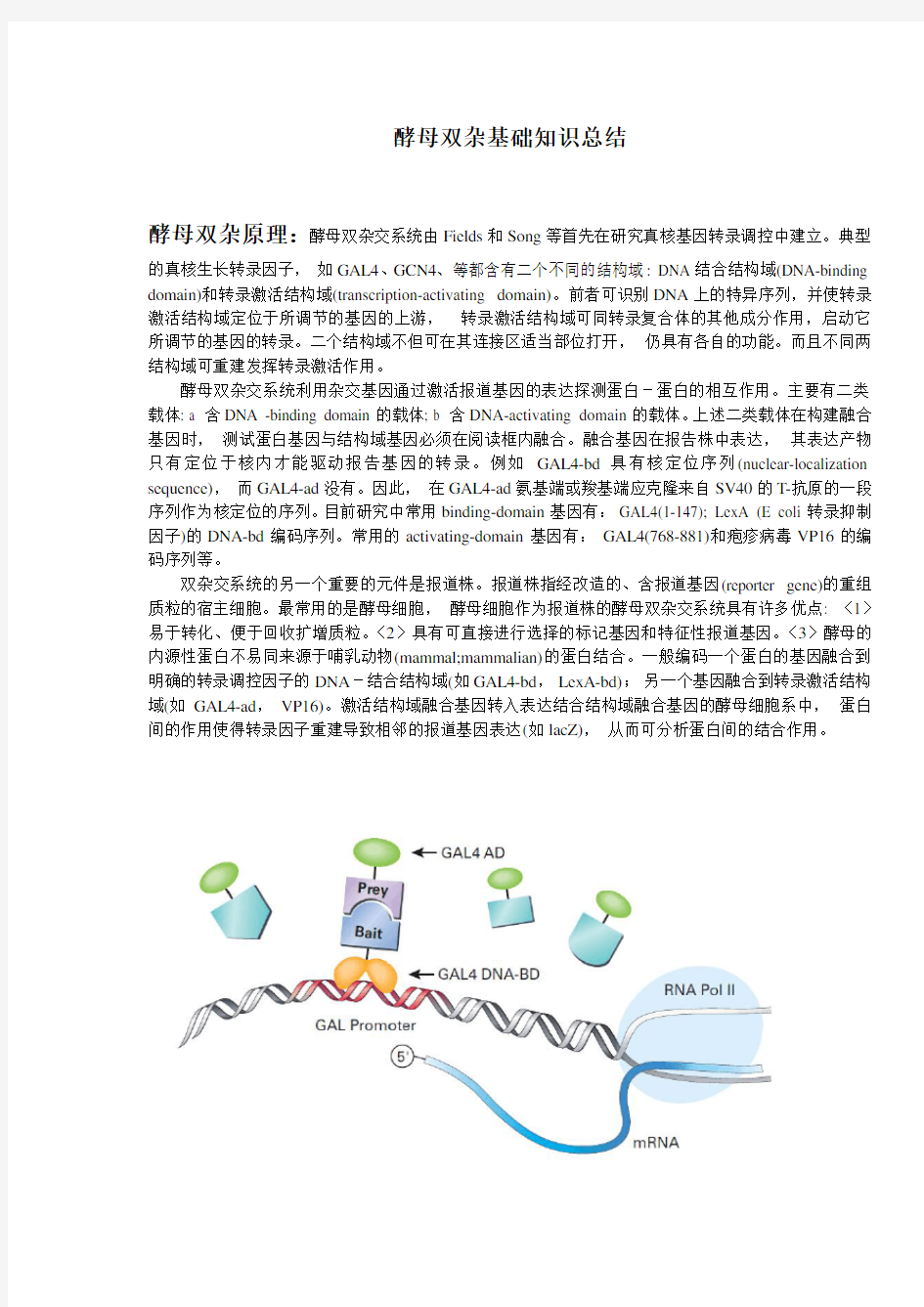
的真核生长转录因子,如GAL4、GCN4、等都含有二个不同的结构域: DNA结合结构域(DNA-binding domain)和转录激活结构域(transcription-activating domain)。前者可识别DNA上的特异序列,并使转录激活结构域定位于所调节的基因的上游,转录激活结构域可同转录复合体的其他成分作用,启动它所调节的基因的转录。二个结构域不但可在其连接区适当部位打开,仍具有各自的功能。而且不同两结构域可重建发挥转录激活作用。
酵母双杂交系统利用杂交基因通过激活报道基因的表达探测蛋白-蛋白的相互作用。主要有二类载体: a 含DNA -binding domain的载体; b 含DNA-activating domain的载体。上述二类载体在构建融合基因时,测试蛋白基因与结构域基因必须在阅读框内融合。融合基因在报告株中表达,其表达产物只有定位于核内才能驱动报告基因的转录。例如GAL4-bd具有核定位序列(nuclear-localization sequence),而GAL4-ad没有。因此,在GAL4-ad氨基端或羧基端应克隆来自SV40的T-抗原的一段序列作为核定位的序列。目前研究中常用binding-domain基因有:GAL4(1-147); LexA (E coli转录抑制因子)的DNA-bd编码序列。常用的activating-domain基因有:GAL4(768-881)和疱疹病毒VP16的编码序列等。
双杂交系统的另一个重要的元件是报道株。报道株指经改造的、含报道基因(reporter gene)的重组质粒的宿主细胞。最常用的是酵母细胞,酵母细胞作为报道株的酵母双杂交系统具有许多优点: 〈1〉易于转化、便于回收扩增质粒。〈2〉具有可直接进行选择的标记基因和特征性报道基因。〈3〉酵母的内源性蛋白不易同来源于哺乳动物(mammal;mammalian)的蛋白结合。一般编码一个蛋白的基因融合到明确的转录调控因子的DNA-结合结构域(如GAL4-bd,LexA-bd);另一个基因融合到转录激活结构域(如GAL4-ad,VP16)。激活结构域融合基因转入表达结合结构域融合基因的酵母细胞系中,蛋白间的作用使得转录因子重建导致相邻的报道基因表达(如lacZ),从而可分析蛋白间的结合作用。
酵母双杂交文库构建基本实验流程
一第一链cDNA合成
1. 准备高质量的Poly A 或总RNA
2. 在微量离心管中混匀以下试剂
RNA 样本(0.025–1.0 μg poly A+或0.10–2.0 μg 总RNA) 1–2 μl
1.0 μl CDS III 1–2 μl
去离子水补齐体系到4.0 μl
注: 对照实验时,请使用1μg的对照RNA。
CDSIII = Oligo-dT;
CDSIII/6 =随机引物
3. 72°C ,孵育2 min。
4. 冰上冷却2 min,然后14000rpm,10s,瞬时离心。
5. 混合以下试剂,将混合液加入Step4中的经过变性且结合有引物的RNA样本中,轻拍混匀。
5X First-Strand 缓冲液 2.0 μl
DTT (100 mM) 1.0 μl
dNTP 混合物(10 mM ) 1.0 μl
SMART MMLV 反转录酶 1.0 μl
6. 42°C,孵育10 min。
7. 加入1 μl SMART III-modified oligo,混匀,42°C,孵育1 h。
8. 75°C ,10 min终止第一链合成反应。
9. 室温冷却,加入1 μl RNA酶H (2U)。
10. 37°C,孵育20 min。
11. 继续进行LD-PCR 扩增。
二第二链cDNA合成(LD-PCR)
1. 建立两管100ul的扩增体系,一个是实验组,另一个是对照组,往实验组离心管中加入以下试剂并混合:
第一链cDNA 2 μl
蒸馏水70 μl
10X Advantage. 2 PCR缓冲液10 μl
50X dNTP 混合物 2 μl
5’PCR 引物 2 μl
3’PCR引物 2 μl
10X溶解液10 μl
50X Advantage 2聚合酶混合物 2 μl
总体积100 μl
2. 扩增程序:
95°C 30 sec
x Cycles
95°C 10s
68°C 6 min
68°C 5min
3. 对每个样品取7 μl PCR产物进行琼脂糖凝胶电泳检测。
三CHROMA SPIN.+TE-400纯化柱纯化ds cDNA
1. 将剩余的93ul的cDNA样品加入CHROMA SPIN TE-400 纯化柱中,颠倒纯化柱数次,重悬胶基质,直至均匀。移除顶端帽子,掰断柱底部的break away,将柱放置在2ml 的收集管中。
2. 700 rpm,5 min离心,丢弃收集管和平衡液,之后让matrix半干。
3. 将将离心柱放在另第二个收集管中,然后,向胶基质平坦的表面上方加入93ul的样本。
4. 700 rpm,5 min离心,样本便流到了收集管中。
5. 将两次的纯化样本收集到一个离心管中,并用乙醇进行沉淀cDNA:
加入1/10体积的3 M 醋酸钠。
加入2.5倍体积的冰乙醇(95–100%)。
–20°C ,1 hr放置沉淀。
14,000 ,室温离心20 min。
弃上清,14,000 rpm瞬时离心,去除残留的上清液。
空气干燥10 min。
6. 20 μl 去离子水重悬cDNA,用于酵母文库构建。
四建立Mate & Plate 文库
1. 遵循酵母转化系统文库规模酵母转化操作,向0.5ml Y187感受态中共转化下列组份:
已纯化的ds cDNA 2–5 μg
pGADT7-Rec (0.5 μg/μl) 6 μl
2. 按照转化操作的第二步,将酵母重悬于15ml 0.9% (w/v) NaCl中。
3.将100 μl 的1/10 、1/100稀释液分别铺板于SD/–Leu 100 mm 固体培养基上,30°C,孵育3–4d,确定文库的库容。
4. 将剩下的悬浮液铺板于150 mm SD/–Leu选择培养基上,每板100 μl 悬浮液,约使用150板,30°
C 孵育3-4d
5. 收获Step4中的转化子:
a. 4°C ,3–4 h冷却培养板。
b. 加入5 ml 的冻存液(YPDA/25%甘油)。
c. 使用无菌的玻璃珠分离菌落(缓缓地摇晃培养板,玻璃珠均匀地滚动于培养基表面将分离菌落,分离后的菌落溶于冻融液中)。
d. 将所有的液体收集于无菌的单管中(收集后的体积要达到0.5 L)。
e.用血球计数板来计算细胞密度,若细胞密度小于<2 x 107/ml,离心来减少悬浮液的体积。
f. 将文库分装到2ml的离心管中(1ml/管),-80℃贮存。
6. 取1ml单管文库进行筛选。
酵母细胞感受态的制备及转化
小样转化文库转化
1 在转化实验的两天前,于YPD培养基的平皿上活化酵母菌株(Y2H/Y187)。
2. 转化前一天,挑取活化的酵母细胞(单克隆)于YPDA液体培养基中,30°C,
200rpm培养8-12h。
3. 取5μl上述酵母细胞液于50ml的YPDA培养基中, 30°C,200rpm培养
8-12h至OD600>1.5。
50ml 150ml 4.将上述酵母细胞液离心后转接到新的YPDA液体培养基中,使OD600=0.2,
30°C 200rpm培养4-6h,使OD600=0.5
1.0
300ml 1L
5. 700g 离心 5分钟收集酵母细胞,用无菌水或TE洗酵母细胞。25-50ml 500ml
6. 再次700g 离心 5分钟。
7. 弃上清,用1.1×TE/LiAc重悬酵母细胞。 1.5ml 8ml
8. 往离心管里加入如下试剂及DNA:
?DNA-BD/baita
?AD/library
?Herring testes carrier DNA 0.1 μg
0.1 μg
0.1 mg
0.2–1.0 mg
0.1–0.5 mg
20 mg
9. 加入感受态细胞,吸打均匀0.1 ml 8 ml
10. 加入PEG/LiAc缓冲液,高速漩涡混合0.6 ml 60 ml
11. 200rpm,30度孵育30min
12. 加入DMSO,轻轻混合70 μl7.0 ml
13. 42度,40min水浴
14. 冰浴1-2min
15. 室温高速离心(14000g) 5s 5min
16. 弃上清,用1×TE重悬,进行涂皿操作0.5ml 10ml
酵母文库的筛选
A 诱饵质粒自激活活性检测和毒性检测
1. 诱饵载体和对照空载体转化酵母Y2H细胞,取100μL涂布SD/-Trp固体培养板,30°C倒
置培养3-4天待菌落长出。
2. 挑(2-3 mm)单克隆至50 ml SD/–Trp 液体培养基中,30°C摇床230-260 rpm,16-24 hr后,菌落
PCR检测目的基因是否转入Y2H中,-70°C保存阳性菌种。
3. 将阳性克隆在SD/-Trp/X-gal(SDO/X) SD/-Trp/X-gal/Aba(SDO/X/A)平板上划线,30°C
倒置培养2天。
4.观察有无蓝色菌斑的出现,菌斑不显蓝色则表明无自我激活活性;若实验菌斑与对照生长状况相同,则诱饵载体对酵母无毒。
注:所需涂的皿及其生长结果如下表:
sample Selective agar plate 2mm colony colour
Y2H[pGBKT7::bait] SDO[SD/-Trp] Yes white
Y2H[pGBKT7::bait] SDO/ X-Gal Yes white
Y2H[pGBKT7::bait] SDO/ X-Gal /Aba NO no
DDO/ X-Gal /Aba NO NO
Y2H[pGBKT7::Lam]+
Y187[pGADT7-T]
DDO/ X-Gal /Aba Yes blue
Y2H[pGBKT7::53]+
Y187[pGADT7-T]
B 结合实验及筛库
1.在冰上冻融1ml文库(酵母菌株≥ 2 x 107cells Y187文库)。
2.在2L三角瓶中,加入5ml 含Y2H酵母菌(≥1 x 109cells/ ml目的蛋白bait)的培养基和上述冻融的1ml文库。
3.加入45ml 2×YPDA/Kan (50μg/ml),30-50rpm,30℃,mating 20-24小时,mating 20小时后可取5μl到显微镜下观察,如还没有结合子存在,再mating 4小时,否则停止mating进入下面操作。
4.准备SD/–Ade/–His/–Leu/–Trp(QDO)培养基平板,约150个(涂皿前应在超净工作台上吹2-3小时)。
5.转移mating体系至2个50ml离心管中,1000g室温离心10分钟,弃上清液。然后再加入2×YPDA洗一次酵母细胞,1000g室温离心10分钟,弃上清液。再用灭菌水清洗酵母细胞,两管合并,1000g室温离心10分钟,弃上清液,加入约10 ml灭菌水重悬酵母细胞。
6.涂皿,每平板加入150μl上述悬浮酵母细胞用玻璃珠涂开至无流动液体。
7.封皿后置于30℃恒温生长箱,生长4-6天后,选取菌斑大于2mm的菌落,在QDO 平板皿上划线,2-3d后长出的菌落可挑出来做插入片段扩增PCR。
8.将(7)中扩增片段大小不同(PCR验证3次,可大大降低假阳性菌落(约40%))的菌落于QDO、QDO/X-Gal和QDO/X-Gal/Aba培养基上划线(可适当增大X-Gal和Aba 的浓度,以提高筛选的严谨性),封皿后置于30℃恒温生长箱,生长2-3d后观察生长状况。若在QDO上生长,QDO/X-Gal菌落变蓝,QDO/X-Gal/Aba不生长,则存在互作。
将存在互作的菌落进行菌落PCR,将PCR产物送去测序,对测序结果进行分析,看是否存在与花粉的发育、能量传递和合成、核酸结合或者剪切、毒性蛋白等相关的基因。9.挑互作酵母菌株至2ml 2×YPDA中,30°C摇床230-260 rpm,16-24 hr,提取酵母质粒,再转入大肠杆菌中扩增含目的基因的载体,测序,到数据库中比对测序结果,标记感兴趣的互作蛋白。
10.阳性克隆验证:抽出互作的酵母质粒后做点对点验证,注意排除自激活情况。
注:点对点验证涂皿情况及其对应生长情况:
sample DDO QDO QDO/ X-Gal QDO/X-Gal/Aba
Y2H[pGBKT7::bait]+
Yes Yes Blue No
Y187[pGADT7::prey]
Yes No No No
Y2H[pGBKT7]+
Y187[pGADT7::prey]
Yes No No No
Y2H[pGBKT7]+
Y187[pGADT7]
Y2H[pGBKT7::Lam]+
Yes No No No
Y187[pGADT7-T]
Yes Yes Blue Yes
Y2H[pGBKT7::53]+
Y187[pGADT7-T]