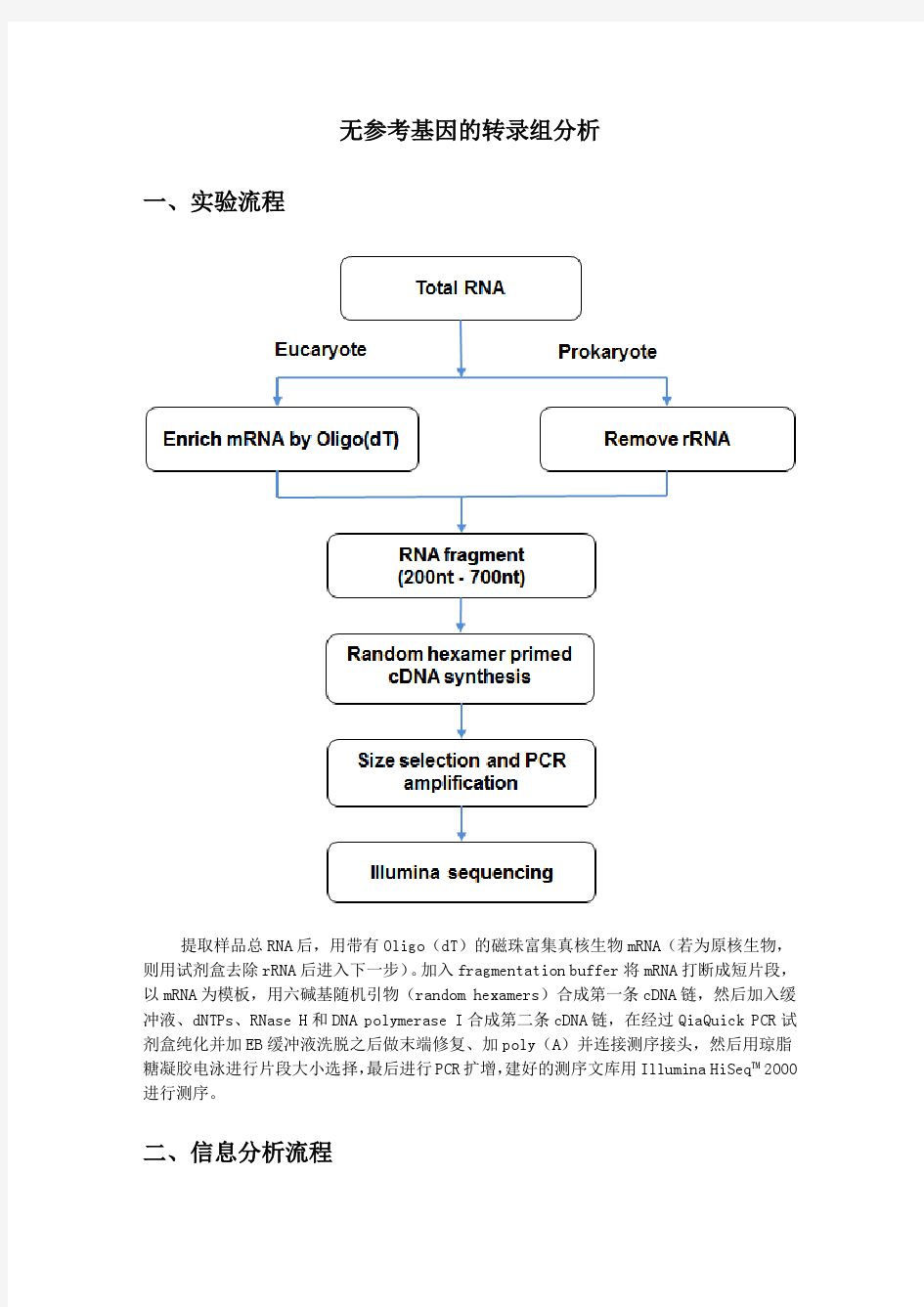
转录组:是一个细胞、组织或有机体在特定条件下表达的一组完整的基因 蛋白质组(Proteomics):指由一个基因组,或一个细胞、组织表达的所有蛋白质. 蛋白质组学的研究内容主要有两方面,一是结构蛋白质组学,二是功能蛋白质组学密码子:mRNA上每3个核苷酸翻译成蛋白质多肽链上的一个氨基酸,这3个核苷酸就称为密码子。 转录:是指拷贝出一条与DNA链序列完全相同的RNA单链的过程。 1大肠杆菌乳糖操纵子包括三个结构基因:Z,Y,A以及一个操纵序列O,一个启动序列P及一个调节基因I等。转录时,RNA聚合酶首先与启动区结合,通过操纵向右转录。转录从启动区又开始,按Z-Y-A得方向进行,每次转录出来的一条mRNA上都带有这三个基因。转录的调控是在启动区和操纵区进行的。 正调控机制:cAMP-CAP复合物与DNA结合改变了这一区段DNA次级结构,促进RNA聚合酶结合区的解链。这可能是cAMP-CAP 通过与RNA聚合酶结合,再与DNA结合,因而促进了RNA聚合酶与启动基因的结合,从而增强了转录。cAMP-CAP复合物的形成取决于细胞内cAMP的浓度,当以葡萄糖为能源时,由于其限制腺苷酸环化酶的活性,AMP不能转化为cAMP,细胞内cAMP的浓度降低,形不成cAMP-CAP复合物,因而乳糖结构基因不被转录。 负调控机制:具有活性的阻遏物只要结合在操纵基因上,就可阻挠RNA聚合酶的转录活动,这是由于P和O位点有一定的重叠序列,O被阻遏物占据后,RNA聚合酶便不能结合到P位点上。阻遏物有无活性又受乳糖这种小分子诱导物的影响。阻遏物与乳糖结合后,由于发生构想变化而失活,不再同操纵基因结合,于是RNA聚合酶便能结合于启动基因,启动基因的表达,使乳糖利用的结构基因转录出相应的mRNA,进而在翻译除蛋白质。在没有到合成这个调节系统中,阻遏蛋白是主要的作用因子,而诱导物可以影响阻遏蛋白的活性,只有阻遏物被诱导失活,结构基因才得以表达。 23. 蛋白质翻译后加工的主要内容包括哪些 a)对真核基因所编码的蛋白质而言,翻译后加工的内容包括: b)除去肽链合成的起始氨基酸或随后几个氨基酸残基; c)分泌蛋白或膜蛋白N-末端信号肽的切除; d)二硫键的形成及氨基酸的共价修饰,包括蛋白N-端氨基酸的豆蔻酰化、蛋白质的
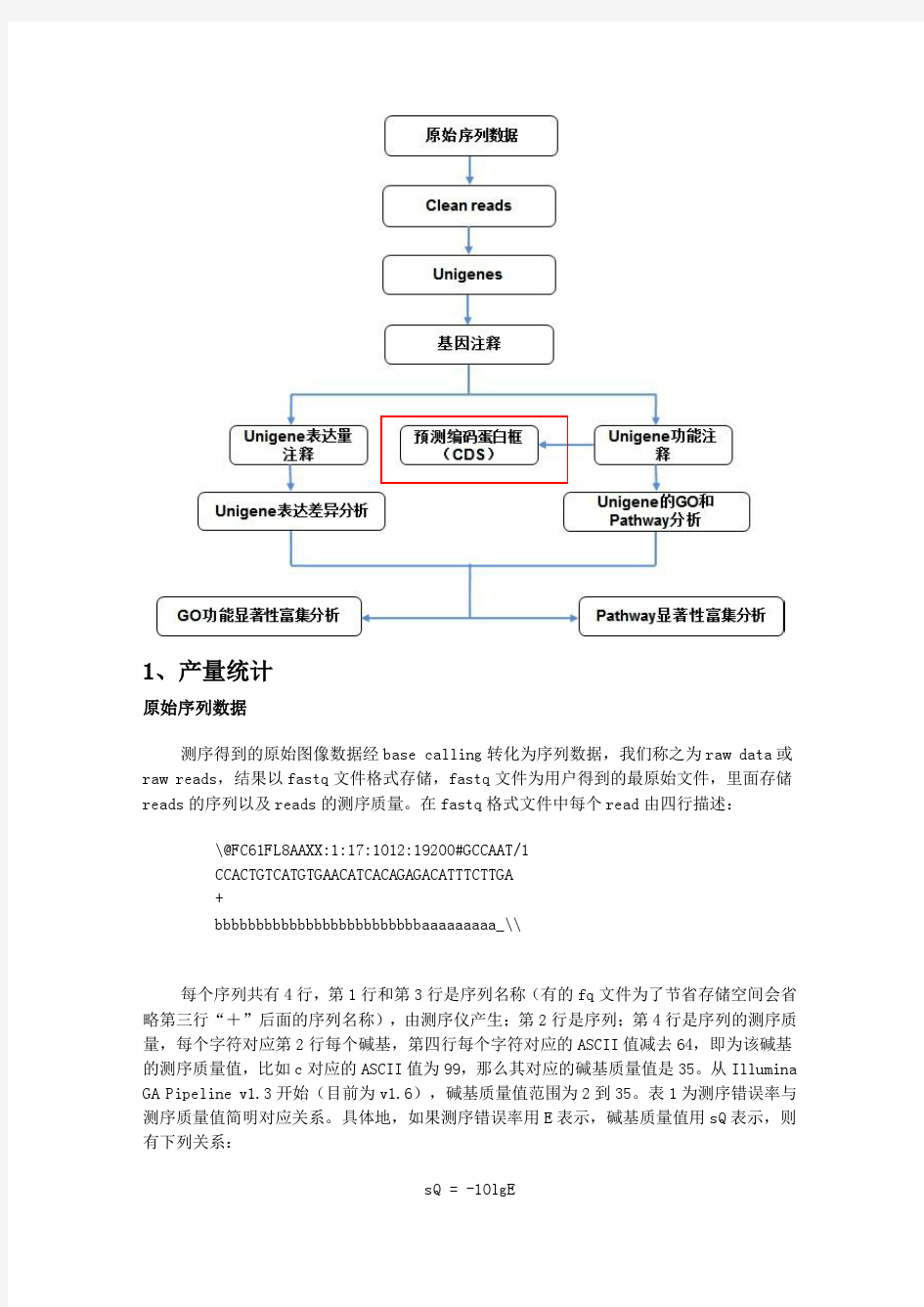
一、生物信息分析流程 获得原始测序序列(Sequenced Reads)后,在有相关物种参考序列或参考基因组的情况下,通过如下流程进行生物信息分析: 二、项目结果说明 1 原始序列数据 高通量测序(如illumina HiSeq TM2000/MiSeq等测序平台)测序得到的原始图像数据文件经碱基识别(Base Calling)分析转化为原始测序序列(Sequenced Reads),我们称之为Raw Data或Raw Reads,结果以FASTQ(简称为fq)文件格式存储,其中包含测序序列(reads)的序列信息以及其对应的测序质量信息。 FASTQ格式文件中每个read由四行描述,如下: @EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG GCTCTTTGCCCTTCTCGTCGAAAATTGTCTCCTCATTCGAAACTTCTCTGT + @@CFFFDEHHHHFIJJJ@FHGIIIEHIIJBHHHIJJEGIIJJIGHIGHCCF 其中第一行以“@”开头,随后为illumina 测序标识符(Sequence Identifiers)和描述文字(选择性部分);第二行是碱基序列;第三行以“+”开头,随后为illumina 测序标识符(选择性部分);第四行是对应序列的测序质量(Cock et al.)。 illumina 测序标识符详细信息如下:
第四行中每个字符对应的ASCII值减去33,即为对应第二行碱基的测序质量值。如果测序错误率用e表示,illumina HiSeq TM2000/MiSeq的碱基质量值用Q phred 表示,则有下列关系: 公式一:Q phred = -10log 10 (e) illumina Casava 1.8版本测序错误率与测序质量值简明对应关系如下: 2 测序数据质量评估 2.1 测序错误率分布检查 每个碱基测序错误率是通过测序Phred数值(Phred score, Q phred )通过公式1转化得到,而Phred 数值是在碱基识别(Base Calling)过程中通过一种预测碱基判别发生错误概率模型计算得到的,对应关系如下表所显示: illumina Casava 1.8版本碱基识别与Phred分值之间的简明对应关系 测序错误率与碱基质量有关,受测序仪本身、测序试剂、样品等多个因素共同影响。对于RNA-seq技术,测序错误率分布具有两个特点: (1)测序错误率会随着测序序列(Sequenced Reads)的长度的增加而升高,这是由于测序过程中化学试剂的消耗而导致的,并且为illumina高通量测序平台都具有的特征(Erlich and Mitra, 2008; Jiang et al.)。 (2)前6个碱基的位置也会发生较高的测序错误率,而这个长度也正好等于在RNA-seq 建库过程中反转录所需要的随机引物的长度。所以推测前6个碱基测序错误率较高的原因为随机引物和RNA模版的不完全结合(Jiang et al.)。测序错误率分布检查用于检测在测序长度范围内,有无异常的碱基位置存在高错误率,比如中间位置的碱基测序错误率显着高于其他位置。一般情况下,每个碱基位置的测序错误率都应该低于0.5%。 图2.1 测序错误率分布图
转录组测序(RNA-seq)技术 转录组是某个物种或者特定细胞类型产生的所有转录本的集合。转录组研究能够从整体水平研究基因功能以及基因结构,揭示特定生物学过程以及疾病发生过程中的分子机理,已广泛应用于基础研究、临床诊断和药物研发等领域。基于Illumina高通量测序平台的转录组测序技术使能够在单核苷酸水平对任意物种的整体转录活动进行检测,在分析转录本的结构和表达水平的同时,还能发现未知转录本和稀有转录本,精确地识别可变剪切位点以及cSNP(编码序列单核苷酸多态性),提供最全面的转录组信息。相对于传统的芯片杂交平台,转录组测序无需预先针对已知序列设计探针,即可对任意物种的整体转录活动进行检测,提供更精确的数字化信号,更高的检测通量以及更广泛的检测范围,是目前深入研究转录组复杂性的强大工具。 技术优势: ?数字化信号:直接测定每个转录本片段序列,单核苷酸分辨率的精确度,同时不存在传统微阵列杂交的荧光模拟信号带来的交叉反应和背景噪音问题。 ?高灵敏度:能够检测到细胞中少至几个拷贝的稀有转录本。 ?任意物种的全基因组分析:无需预先设计特异性探针,因此无需了解物种基因信息,能够直接对任何物种进行转录组分析。同时能够检测未知基因,发现新的转录本,并精确地识别可变剪切位点及cSNP,UTR区域。 ?更广的检测范围:高于6个数量级的动态检测范围,能够同时鉴定和定量稀有转录本和正常转录本。 应用领域:转录本结构研究(基因边界鉴定、可变剪切研究等),转录本变异研究(如基因融合、编码区SNP研究),非编码区域功能研究(Non-coding RNA研究、microRNA前体研究等),基因表达水平研究以及全新转录本发现。 图1 RNA-seq获得的数据能够进行全面的数据挖掘,既能够进行基因结构分析,鉴定UTR、可变剪切位点,也能够发现新的转录本及非编码RNA,比较样本间的表达水平差异
生物基因组非蛋白质编码转录组学及研究进展 姜 宁1 陈启军 2 1.中国医学科学院 吉林大学人兽共患病联合研究中心人兽共患病研究教育部重点实验室,长春130062 2.中国医学科学院病原生物学研究所,北京100730 收稿日期:2009 9 13 修回日期:2009 12 1联系作者:陈启军,教授,cq@j jl https://www.doczj.com/doc/3416265530.html, .cn 。 摘 要 RNA 转录组学和功能组学的研究是目前生命科学领域的重要研究方向。生命的中心法则(由DNA 转录RNA,再由后者翻译成行使各种功能的蛋白质)因调控RNA 分子的发现而进一步得到扩展。最近的大量研究发现,自基因组中非蛋白质编码区转录的RNA 分子具有重要的调控功能,即转录后的调控功能。在这些RNA 分子中,内源性小干扰RNA 分子、m icroRNA 及pi w i RNA 等的功能逐渐被揭示。本文对目前有关RNA 转录组学研究进展做一简要综述。 关键词:RNA 转录组 小RNA si R NA m i R NA pi R NA 中图分类号:Q7 文献标识码:A 文章编号:1009 2412(2009)06 0015 05 一、引 言 生物物种遗传物质的组成随着物种进化程度的 提高而逐渐趋于复杂。然而随着大规模基因组测序的完成,人们发现很多生物(包括小鼠和人)遗传物质组成的主要差异不是在蛋白质编码区而是在基因组中的非编码(non cod i ng )区。生物物种的种源进化程度越高,其基因组中非蛋白质编码序列的组成比例越高[1],如人类基因组中编码蛋白质的DNA 只占基因组的2%左右。长期以来,对基因组序列的研究多集中在对编码区的分析上(如基因的序列组成,编码蛋白质的表达、功能及调控规律等)。由于非编码区的序列多含有一些假基因(ps eudo genes)、转座 子(trans poson 或trans posab le ele m ents)及大量的内含子和重复序列,其潜在的功能一直为研究者们所忽视。多年来人们一直将基因组中非编码序列认为是生物进化过程中形成的垃圾成分(junk DNA )[2]。然而,随着大规模转录组学(transcripto m ics)研究的进行,发现基因组中绝大部分DNA 在细胞活动过程中都是被转录成RNA 的[3],如人类基因组DNA 有93%以上都被转录成RNA,小鼠基因组的转录部分也达到63%以上[3]。这些RNA 有的呈单链存在,有的以双链形式存在。对RNA 转录组的研究经历了小RNA 的发现、大规模RNA 转录组的测定到目前的RNA 调控功能的分析和确定等阶段[3 8] 。RNA 转录 组学和功能组学的研究是目前生命科学领域的重要 研究方向。 二、基因组中非编码区转录产生的 RNA 分子种类及功能 根据RNA 片段长度的不同,自基因组中转录的 RNA 分子包括短片段RNA (s hort RNA )和长片段RNA (l ong RNA )[1,7,9,10]。短片段RNA 分子主要包括反式剪切引导RNA (trans splicing leader RNA,S L RNA )、m i cro RNA (m i R NA )、内源性小干扰RNA (en dogenous s m all i nterferi ng RNA,si R NA )、p i w i 蛋白质 结合RNA (p i w i RNA, pi RNA )和一些编码寡肽的小 mRNA 分子[11]。内源性小RNA (endogenous s m all non cod i ng RNA, s n RNA)是一类从基因组中非蛋白 质编码区转录而来的小RNA 分子。目前对内源性s nRNA 的研究主要集中在对S L RNA 、si R NA 和m i R NA 等的发现及功能分析方面。这些小RNA 主要通过影响mRNA 的成熟过程及稳定性进而调节转录因子或其它功能蛋白质的表达和发挥转录后的基因调控功能(post transcri pt i ona l gene regulat i on ,PTGR )。long RNA 主要指mRNA 前体(hnRNA )、mRNA 和一些不编码任何蛋白质的长的单链或双链RNA 片段。
《基因组学与蛋白质组学》课程教学大纲 学时: 40 学分:2.5 理论学时: 40 实验学时:0 面向专业:生物科学、生物技 术课程代码:B7700005先开课程:生物化学、分子生物 学课程性质:必修/选修执笔人:朱新 产审定人: 第一部分:理论教学部分 一、课程的性质、目的和任务 《基因组学与蛋白质组学》是随着生物化学、分子生物学、结构生物学、晶体学和计算机技术等的迅猛发展而诞生的,是融合了生物信息学、计算机辅助设计等多学科而发展起来的新兴研究领域。是当今生命科学研究的热点与前沿领域。由于基因组学与蛋白质组学学科的边缘性,所以本课程在介绍基因组学与蛋白质组学基本基本技术和原理的同时,兼顾学科发展动向,讲授基因组与蛋白组学中的热点和最新进展,旨在使学生了解现代基因组学与蛋白质组学理论的新进展并为相关学科提供知识和技术。 二、课程的目的与教学要求 通过本课程的学习,使学生掌握基因组学与蛋白质组学的基本理论、基础知识、主要研究方法和技术以及生物信息学和现代生物技术在基因组学与蛋白质组学上的应用及典型研究实例,熟悉从事基因组学与蛋白质组学的重要方法和途
径。努力培养学生具有科学思维方式、启发学生科学思维能力和勇于探索,善于思考、分析问题的能力,激发学生的学习热情,并通过学习提高自学能力、独立思考能力以及科研实践能力,为将来从事蛋白质的研究奠定坚实的理论和实践基础。 三、教学内容与课时分配 第一篇基因组学
第一章绪论(1学时) 第一节基因组学的研究对象与任务; 第二节基因组学发展的历程; 第三节基因组学的分子基础; 第四节基因组学的应用前景。 本章重点: 1. 基因组学的概念及主要任务; 2. 基因组学的研究对象。 本章难点: 1.基因组学的应用及发展趋势; 2.基因组学与生物的遗传改良、人类健康及生物进化。建议教学方法:课堂讲授和讨论 思考题: 查阅有关资料,了解基因组学的应用发展。 第二章人类基因组计划(1学时) 第一节人类基因组计划的诞生; 第二节人类基因组研究的竞赛; 第三节人类基因组测序存在的缺口; 第四节人类基因组中的非编码成分; 第五节人类基因组的概观; 第六节人类基因组多样性计划。 本章重点: 1. 人类基因组的研究; 2. 人类基因组多样性。 本章难点: 人类基因组序列的诠释。 建议教学方法:课堂讲授和讨论 思考题:
转录组测序结题报告 1.mRNA纯化: 抽提得到的总RNA首先利用10U的DNaseI(Ambion,美国)在37℃消化1小时;然后利用Micropoly(A)PuristTM mRNA purification kit(Ambion,美国),进行mRNA纯化:把RNA稀释到250μl的体积,按照Kit的操作步骤(Cat.No:
1919)进行;最后得到的mRNA用100μl预热的THE缓冲液洗脱,利用NanoDrop 进行定量。 2.cDNA合成: cDNA合成是在Ng等2005年发表的方法基础上改进而成(文献1,图1)。第一链cDNA合成利用GsuI-oligo dT作为反转录引物,10μg的mRNA作为模板,用1000 单位的Superscript II reverse transcriptase (Invitrogen,美国)在42℃作用1小时完成;随后利用NaIO4(Sigma,美国)氧化mRNA的5’帽子结构,并连接生物素;通过Dynal M280磁珠(Invitrogen,美国)筛选连接了生物素的mRNA/cDNA,并通过碱裂解释放第一链cDNA;然后通过DNA ligase(TaKaRa,日本)在第一链cDNA的5’末端加上接头,然后通过Ex Taq polymerase (TaKaRa,日本)合成第二链cDNA。最后通过GsuI酶切去除polyA和5’端接头。 图1. 全长cDNA合成示意图 3.cDNA测序: 合成的cDNA利用超声仪(Fisher)打断到300-500bp的范围,利用Ampure beads(Agencourt,美国)进行纯化。随后纯化的cDNA利用TruSeq TM DNA XXmple Prep Kit – Set A (illumina,美国)制备文库,并利用TruSeq PE Cluster Kit (illumina,美国)进行扩增。最后在illumina机器上进行测序反应。 测序得到的数据统计见表1. 表1. Solexa测序统计 样品对照 1 2
RNA-Seq名词解释 1.index 测序的标签,用于测定混合样本,通过每个样本添加的不同标签进行数据区分,鉴别测序样品。 2.碱基质量值 (Quality Score或Q-score)是碱基识别(Base Calling)出错的概率的整数映射。碱基质量值越高 表明碱基识别越可靠,碱基测错的可能性越小。 3.Q30 碱基质量值为Q30代表碱基的精确度在99.9%。 4.FPKM(Fragments Per Kilobase of transcript per Million fragments mapped) 每1百万个map上的reads中map到外显子的每1K个碱基上的fragment个数。计算公式为 公式中,cDNA Fragments 表示比对到某一转录本上的片段数目,即双端Reads数目;Mapped Reads(Millions)表示Mapped Reads总数, 以10为单位;Transcript Length(kb):转录本长度,以kb个碱基为单位。 5.FC(Fold Change) 即差异表达倍数。 6.FDR(False Discovery Rate) 即错误发现率,定义为在多重假设检验过程中,错误拒绝(拒绝真的原(零)假设)的个数占所有被拒绝 的原假设个数的比例的期望值。通过控制FDR来决定P值的阈值。 7.P值(P-value) 即概率,反映某一事件发生的可能性大小。统计学根据显著性检验方法所得到的P 值,一般以P<0.05 为显著,P<0.01为非常显著,其含义是样本间的差异由抽样误差所致的概率小于0.05或0.01。 8.可变剪接(Alternative splicing)
(内部资料,请勿外传) 动植物转录组 (Transcriptome ) 产品说明书 科技服务体系 动植物研究方向
版本信息: 2011年07月08日
目录 1产品概述 (1) 1.1 什么是转录组测序 (1) 1.2 转录组测序的产品功能 (1) 1.3 转录组测序产品优势 (1) 1.4 转录组测序产品发展史 (1) 1.5 项目执行时间 (3) 1.6 产品交付结果 (3) 2转录组测序研究方法 (4) 2.1 产品策略 (4) 2.2 样品准备 (5) 2.2.1 RNA样品要求 (5) 2.2.2 RNA样品送样标准 (6) 2.2.3 RNA提取的组织用量建议 (6) 2.3 样品运输要求 (7) 2.3.1 样品包装 (7) 2.3.2 样品标识 (8) 2.3.3 样品运输条件 (8) 2.4 文库的构建及测序 (9) 2.4.1 实验流程 (9) 2.4.2 测序及数据处理 (10) 2.5 转录组生物信息学分析 (10) 2.5.1 没有参考序列的转录组De novo (10) 2.5.2 有参考序列的转录组Re-sequencing (18) 2.5.3 参考文献 (24) 3成功案例 (25)
3.1 华大成功案例 (25) 3.2 相关文献解读 (26)
1产品概述 1.1什么是转录组测序? 转录组测序的研究对象为特定细胞在某一功能状态下所能转录出来的所有RNA的总和,包括mRNA和非编码RNA。转录组测序是指用新一代高通量测序技术对物种或者组织的转录本进行测序并得到相关的转录本信息。 1.2转录组测序的产品功能 1.获得物种或者组织的转录本信息; 2.得到转录本上基因的相关信息,如:基因结构,功能等; 3.发现新的基因; 4.基因结构优化; 5.发现可变剪切; 6.发现基因融合; 7.基因表达差异分析。 1.3转录组测序产品优势 覆盖度高:检测信号是数字信号,几乎覆盖所有转录本; 检测精度高:几十到数十万个拷贝精确计数; 分辨率高:可以检测到单碱基差异,基因家族中相似基因及可变剪切造成的不同转录本的表达; 完成速度快:整个项目周期只需要50个工作日时间; 成本低:基本上每个实验室可以承担相关研究经费。 1.4转录组测序产品发展史 转录组的研究手段大体包括:EST序列构建及研究,芯片研究,运用第二代测序技术研究等。EST是从一个随机选择的cDNA 克隆进行5’端和3’端单一次sanger测序获得的短的cDNA 部分序列,代表一个完整基因的一小部分,在
【摘要】基因组相对较稳定,而且各种细胞或生物体的基因组结构有许多基本相似的特征;蛋白质组是动态的,随内外界刺激而变化。对蛋白质组的研究可以使我们更容易接近对生命过程的认识。蛋白质组学是在细胞的整体蛋白质水平上进行研究、从蛋白质整体活动的角度来认识生命活动规律的一门新学科,简要介绍功能基因组学和蛋白质组学的科学背景、概念及其应用。 【关键词】基因组;功能基因组学;蛋白质组学; 一、基因组及基因组学的概念 基因组(genome)一词系由德国汉堡大学H.威克勒教授于1920年首创,用以表示真核生物从其亲代所继承的单套染色体,或称染色体组。更准确地说,基因组是指生物的整套染色体所含有的全部DNA序列。由于在真核细胞的线粒体和植物的叶绿体中也发现存在遗传物质,因此又将线粒体或叶绿体所携带的遗传物质称为线粒体基因组或叶绿体基因组。原核生物基因组则包括细胞内的染色体和质粒DNA。此外非独立生命形态的病毒颗粒也携带遗传物质,称为病毒基因组。所有生命都具有指令其生长与发育,维持其结构与功能所必需的遗传信息,本书中将生物所具有的携带遗传信息的遗传物质总和称为基因组。[1] 基因组学(genomic)一词系由T.罗德里克(T.Roderick)于1986年首创,用于概括涉及基因组作图、测序和整个基因组功能分析的遗传学学科分支,并已用来命名一个学术刊物Genomics。基因组学是伴随人类基因组计划的实施而形成的一个全新的生命科学领域。[1] 基因组学与传统遗传学其他学科的差别在于,基因组学是在全基因组范围研究基因的结构、组成、功能及其进化,因而涉及大范围高通量收集和分析有关基因组DNA的序列组成,染色体分子水平的结构特征,全基因组的基因数目、功能和分类,基因组水平的基因表达与调控以及不同物种之间基因组的进化关系。基因组学的研究方法、技术和路线有许多不同于传统遗传学的特点,各相关领域的研究仍处于迅速发展和不断完善的过程中。 基因组学的主要工具和方法包括:生物信息学,遗传分析,基因表达测量和基因功能鉴定。 二、功能基因组学的概念及应用
基于基因组学与转录组学的胡桃科植物系统进化及群体遗传学 研究 胡桃科(Juglandaceae)隶属于壳斗目(Fagales),是世界重要的经济树种,具有重要的材用、食用、药用、生态和艺术价值。本研究以胡桃科植物为研究对象,采用高通量测序技术结合生物信息学、进化生物学及群体遗传学等方法,对胡桃科物种进行如下分析:首先,利用群体基因组学数据对该科中最重要的经济树种胡桃属(Juglans)植物进行研究,从多角度揭示胡桃属系统发育关系、物种形成机制以及该属物种复杂的群体动态历史。 其次,本研究利用叶绿体基因组数据阐明胡桃科的系统发育关系、揭示其进化起源中心以及多样化历史,结合化石证据进一步确定胡桃科的在时间尺度上的进化历程。主要结果如下:(1)中国胡桃属植物包括以下5个物种:核桃、铁核桃、野核桃、麻核桃和核桃楸。 首先,基于IlluminaMiseq测序平台首次对胡桃科中核桃的叶绿体DNA进行高通量测序。利用生物信息学方法获得了完整的核桃叶绿体参考基因组序列(160,367 bp)。 对参考基因组序列进行注释,发现其共有137个基因,包括86个蛋白编码基因,3个假基因(2个ycf15和1个infA),40个tRNA基因,8个rRNA基因。其次,由于缺乏丰富的分子标记,中国胡桃属植物5个物种间系统发育关系仍然没有彻底被解决。 本研究利用高通量测序平台Illumina Hiseq对中国5个胡桃属的叶绿体DNA 进行测序,通过上述部分构建的参考叶绿体基因组,进行5个胡桃属叶绿体基因组比较研究。基于比较结果,共鉴定了胡桃属植物叶绿体序列中大量的SNPs和
Indels变异位点,以及简单重复序列和大片段重复序列。 同时,利用叶绿体基因组、蛋白编码基因和非编码区序列三组数据对5个胡桃属进行系统发育分析,结果与形态学的分组高度一致,分为核桃组和核桃楸组。本研究中开展的胡桃属植物叶绿体基因组测序分析将为进一步研究胡桃属的种间杂交、系统进化和群体历史提供可用的遗传资源。 (2)胡桃属植物比较转录组学以及跨物种EST-SSRs分子标记开发可以为后续研究该属物种群体适应性分化研究提供有效的基因组资源。利用Illumina Hiseq测序平台分别对中国5个胡桃属植物,即核桃、野核桃、核桃楸、麻核桃和铁核桃的不同组织(叶片、幼果、雌花、雄花)RNA等量混合后进行转录组测序。 本研究共产生16,811,432-49,929,297 个高质量的 reads,通过 de novo 组装得到 83,112-103,167 个unigenes序列,鉴定出9,216-9,389个核心单拷贝直系同源基因。同时,随机选择96对EST-SSRs分子标记在5个胡桃属物种中进行通用性和多态性检测。 此外,基于467个单拷贝直系同源基因对7个胡桃属植物(核桃、铁核桃、野核桃、核桃楸、麻核桃、美国白核桃和黑核桃)和3个外类群(山核桃、板栗和夏栎)进行系统发育分析,结果表明基因树和物种树系统发育关系一致。基于胡桃属的叶绿体基因组和单拷贝直系同源基因序列分别构建系统发育树的结果表明,美国白核桃和中国特有种麻核桃的系统位置存在分歧,近缘种种间杂交和叶绿体捕获可能是导致胡桃属物种核基因组与叶绿体基因组系统发育关系分歧的原因。 (3)由于胡桃属植物的天然分布是典型的北半球间断分布,而成为东亚-北美生物地理分布模式的研究热点。有限的分子标记不能很好的解决胡桃属的系统发育关系和生物地理分布模式。
蛋白质组与转录组比较关联分析方案一.概述 1.研究背景 生命体是一个多层次,多功能的复杂结构体系,高通量技术的发展积累了大量的组学数据,这使得由精细的分解研究转向系统的整体研究成为可能,整合多组学数据能够实现对生物系统的全面了解。当部分层面上的研究都逐渐走向完善的时候,从部分到整体就是一种必然发展趋势。 相关研究表明,基因表达不仅仅是从转录组到蛋白质组的单向流动,而是两者的相互连接。对这种功能调控的了解通常只限于特殊的信号途径,要了解转录组和蛋白质组之间的相互调控作用,就需要对RNA和蛋白质的表达进行同步监测。 正如RNA可作为部分生物学功能的酶反应的效益物一样,蛋白质也是大多数生物学功能的效益物。因此,蛋白质水平广泛的基因组分析是基因表达更直接的反映。质谱技术的发展,使得定量的蛋白组学研究成为可能。然而,当细胞适应了转录水平、转录后(如mRNA的剪接)、翻译后(蛋白降解和输出)的精细调控机制后,转录物和蛋白质丰度测量结果可能会不一致。因此,定量的转录物和蛋白质丰度测量可作为相互的标准,为高通量分析得出的基因表达数据做出合理的解释。正如蛋白质和RNA之间类似点可以增加我们对新的生物标记的信任度一样,差异也能暗示我们“其他的转录后调控结合点可作为重要的调控研究靶点”。 在蛋白组学分析过程中,一些研究选择了双向凝胶电泳(2一DE)分析蛋白质混合物。要么是对不同的凝胶染色,要么是让不同的细胞与不同的染料相结合,通过斑点染色亮度可以看到蛋白质的亮度。随后用质谱仪对分离出的定量凝较斑点进行鉴定,与转录组学分析不同的是,双向凝胶电泳分析的鉴定结果与定量分析是散耦合(de一coupled)。 液相色谱法(LC)是作为一种替代2一DE的蛋白质分析方法而出现的。LC一MS分析是典型的“自下而上(Bottom一up)”分析方法,通常要用特异的蛋白酶(如胰蛋白酶)将蛋白质消化为肽段。与2一DE不同,LC一MS对肽的定量和鉴定是同时进行的,可以选择定量的MS峰(m/z)用于鉴定,通过肽段的信息推测对应蛋白质的定量信息。 虽然采用的技术不同,迄今为止公开发表的整合分析文章中,都指出了转录组学和蛋白组学的重要性。转录组学或蛋白组学通常只考虑调节系统和分解作用平衡态的净效应,实际上,出现的不一致性只是合成与降解两种替换过程中的一种反映。科学家可能对变化过程中的机制更感兴趣。 正如中心法则预测的那样,在转录物和蛋白质水平,如果只能通过严格的转录调控去控制蛋白质的合成,细胞是不太可能选择精细调节机制的。当点对点进行比较时,蛋白质和转录物之间的一致性通常很弱,这些观察说明了“从个体基
转录组结果解读 转录调控研究部 北京诺禾致源科技股份有限公司
OUTLINE 简介 实验部分 生物信息分析
概述 1 转录组是指特定组织或细胞在某个时间或某个状态下转录出来的所有RNA的总和,主要包括mRNA和非编码RNA。 转录组研究是研究基因功能和结构的基础,对生物体的发育和疾病的发生具有重要作用。 RNA-seq技术流程主要包含两个部分,建库测序和数据分析。
2 实验部分(RNA检测、建库、测序)) ?琼脂糖凝胶电泳:分析样品RNA 完整性及是否存在杂质污染。 ?NanoPhotometer spectrophotometer:检测RNA 纯度(OD260/280及 OD260/230比值)。 ?Agilent 2100 bioanalyzer:精 确检测RNA完整性。 链特异性文库优势: 相同数据量下可获取更多有效 信息;能获得更精准的基因定 量、定位与注释信息
5 ?1、一般动物样品会有三条带:28S 、18S 、5S ,如果提取过程经过过柱处理或者 利用CTAB+LiCl 方法提取,5S 可能较暗或者没有。 ?昆虫或者软体动物等样品只有1条比较明显的带,例如:牡蛎、果蝇、螨虫、蝗 虫、蚊、蚕等 ?2、植物样品有三条带:25S 、18S 、5S ,有些特殊物种或部位可能本身含条带比 较多,如果条带清晰,也可初步判定合格 ?3.原核生物中主要有5S 、16S 、23S rRNA 叶片小 鼠蚊动物植物原核
RIN 5RIN 7RIN 8RIN 9RIN 4RIN 6RIN 10RIN 2RIN 1 RIN 值范围示意图
NHXXXXXX_species转录组生物信息分析结题报告建库测序流程 Total RNA样品检测 文库构建 库检 上机测序 生物信息分析流程 结果展示及说明 原始序列数据 测序数据质量评估 参考序列比对分析 可变剪切分析 新转录本预测 SNP和InDel分析 基因表达水平分析 RNA-seq整体质量评估 基因差异表达分析 差异基因GO富集分析 差异基因KEGG富集分析 差异基因蛋白互作网络分析 参考文献 附录 文件目录列表 软件列表 Methods英文版 备注
一、建库测序流程 从RNA样品到最终数据获得,样品检测、建库、测序每一个环节都会对数据质量和数量产生影响,而数据质量又会直接影响后续信息分析的结果。为了从源头上保证测序数据的准确性、可靠性,诺禾致源对样品检测、建库、测序每一个生产步骤都严格把控,从根本上确保了高质量数据的产出。流程图如下:
1 Total RNA样品检测 诺禾致源对RNA样品的检测主要包括4种方法: (1) 琼脂糖凝胶电泳分析RNA降解程度以及是否有污染 (2) Nanodrop检测RNA的纯度(OD260/280比值) (3) Qubit对RNA浓度进行精确定量 (4) Agilent 2100精确检测RNA的完整性 2 文库构建 样品检测合格后,用带有Oligo(dT)的磁珠富集真核生物mRNA(若为原核生物,则通过试剂盒去除rRNA来富集mRNA)。随后加入fragmentation buffer将mRNA打断成短片段,以mRNA为模板,用六碱基随机引物(random hexamers)合成一链cDNA,然后加入缓冲液、dNTPs和DNA polymerase I合成二链cDNA,随后利用AMPure XP beads纯化双链cDNA。纯化的双链cDNA再进行末端修复、加A尾并连接测序接头,然后用AMPure XP beads进行片段大小选择,最后进行PCR富集得到最终的cDNA文库。构建原理图如下: 3 库检 文库构建完成后,先使用Qubit2.0进行初步定量,稀释文库至1ng/ul,随后使用Agilent 2100对文库的insert size进行检测,insert size符合预期后,使用Q-PCR方法对文库的有效浓度进行准确定量(文库有效浓度>2nM),以保证文库质量。 4 上机测序 库检合格后,把不同文库按照有效浓度及目标下机数据量的需求pooling后进行HiSeq/MiSeq测序。
转录组分析 研究背景: RNA-Seq是通过结合实验和计算方法来鉴定生物样品中RNA序列的种类和丰度的一种技术。通过RNA-seq,我们就能够确定单链RNA分子中ATCG的顺序。整个过程主要包括:从细胞或组织中提取RNA分子、文库的构建以及后继的生物信息学数据分析。RNA-Seq技术具有许多早期研究方法(如:微阵列)所不具备的优点,如:RNA-Seq平台的高通量、新技术所带来的高灵敏度、发现新转录本、新基因模型以及非编码RNA的能力等。 RNA-Seq技术的到来,使人们认识到,无论是单细胞模式生物还是人类,我们对其转录组的认知异常匮乏。而RNA-Seq产生的新的数据,则可以帮助我们发现基因结构上的巨大差异、鉴定出新的转录本以及能够对small non-coding RNA和lncRNAs有着更好的了解。而且随着测序花费的降低,RNA-Seq的优势体现的更加明显。 服务流程: 样品选取
mRNA片段化 cDNA合成 末端修复、加polyA、加接头,PCR扩增 数据分析 测序方案: 内容:TotalRNA检测,普通转录组文库构建及测序及信息分析。测序方式:HiseqPE125。 项目周期:有参45天,无参50天。 分析内容: 无参考基因组: 1.1质量控制 1.11评估碱基质量 1.12过滤低质量reads 1.13 去掉低质量碱基和接头序列 1.14 统计N比例和reads长度 1.15 统计GC含量和reads重复度 1.2 Reads的从头比对组装
1.4基因表达差异分析 1.41 统计基因在不同条件下的差异表达情况 1.5差异基因富集分析 1.51 通过GO、KEGG对差异基因进行功能富集分析 1.6差异表达基因的蛋白质互作网络分析 1.7SNV/Indel分析 1.8样本间相关性分析 有参考基因组: 2.1质量控制(同无参) 2.2 Reads比对组装 2.22 统计reads与参考基因组比对情况 2.22 分析对插入、删除和连接体情况 2.23 统计转录本在参考基因组上位置、长度和覆盖度情况 2.3基因表达差异分析 2.4差异基因富集分析 2.5差异表达基因的蛋白质互作网络分析 2.6新转录本预测 2.7 SNV/Indel分析 2.8 UTR分析 2.9可变剪接分析 3.0 Non-coding RNA分析 3.1样本相关性分析 案例解读: 案例:通过poly(A)+ RNA-Seq分析Drosophila melanogaster转录组的动态性 本项研究通过poly(A)+ RNA-Seq技术对果蝇的细胞系进行测序,鉴定出一批通过替换启动子和RNA剪接来转录出大量转录本的神经特异性基因。通过后继分析还发现,对于RNA剪接变化,组织间的差异要远远大于发育阶段间的差异。另外,发现性腺表达了成百上千的未知的蛋白编码和lncRNAs,其中一些甚至是反义转录的。显示了果蝇转录组的动态性和多样性。 小部分的基因(0.2%)编码出大部分的转录本。
转录组测序结题报告 篇一:转录组测序问题集锦 转录组测序问题集锦 转录组是某个物种或者特定细胞类型产生的所有转录本的集合,转录组测序(RNA-seq) 是最近发展起来的利用深度测序技术进行转录分析的方法,可以对全转录组进行系统的研究。 Roche GS FLX Titanium 、Illumina Solexa GA IIx和AB SOLID 4均可以对转录组进行测序, Roche GS FLX Titanium与Illumina Solexa GA IIx和AB SOLID 4相比,拥有更长的读长和较小的数据量,适用于表达量较高基因的RNA全长测序。但是对低表达丰度的基因,可能需要多次测序才能得到足够的数据,成本比较高,而Illumina Solexa GA IIx和AB SOLID 4数据读取量大,能够得到较高的覆盖率,可以较好的降低成本。若是位置基因组序列的物种,则Roche GS FLX Titanium测序更有优势,其较长的读长便于拼接,获得更好的转录本数据。 转录组测序可以供研究者在转录本结构研究(基因边界鉴定、可变剪切研究等),转录本变异研究(如基因融合、编码区 SNP研究),非编码区域功能研究(Non-coding RNA
研究、miRNA前体研究等),基因表达水平研究以及全新转录本发现等方面进行深入研究。 研究转录组的方法有哪些? 目前研究转录组的方法主要三种,基于杂交技术的cDNA 芯片和寡聚核苷酸芯片,基于sanger测序法的SAGE (serial analysis of gene expression)、LongSAGE和MPSS(massively parallel signature sequencing),基于第二代测序技术的转录组测序,又称为RNA-Seq。 转录组测序比其他研究方法有哪些优势? (1)可以直接测定每个转录本片段序列、单核苷酸分辨率的准确度,同时不存在传统微阵列杂交的荧光模拟信号带来的交叉反应和背景噪音问题; (2)灵敏度高,可以检测细胞中少至几个拷贝的稀有转录本; (3)可以对任意物种进行全基因组分析,无需预先设计特异性探针,因此无需了解物种基因信息,能够直接对任何物种进行转录组分析,同时能够检测未知基因,发现新的转录本,并准确地识别可变剪切位点及cSNP,UTR区域。 (4)检测范围广,高于6个数量级的动态检测范围,能够同时鉴定和定量稀有转录本和正常转录本。