
氨噻肟乙酸苯并噻唑硫酯的合成
周鸿娟1, 张丽君2, 傅德才3
(1.河北师范大学实验中心,河北石家庄050016;2.河北师范大学化学学院,河北石家庄050016;
3.河北科技大学化学工程与制药学院,河北石家庄050018)
摘 要:改进第三代头孢菌素类抗生素中间体氨噻肟乙酸苯并噻唑硫酯的合成方法。采用二氯甲烷和甲苯(体积比1∶1)的混合溶剂代替单一溶剂二氯甲烷,反应可在室温下进行,不需要进一步的精制,提高了产品品质,收率达到85%。中试结果证明:产品品质稳定,重现性好,工业生产过程易于控制。
关键词:合成;氨噻肟乙酸苯并噻唑硫酯;抗生素;头孢菌素中图分类号:O 626 文献标识码:A 文章编号:036726358(2002)1220647203
Syn thesis of 22(22Am ino 242th iazo lyl )222(Z )2m ethoxyi m inoacetic
A cid 22
B enzth iazo lyl T h i oester
ZHOU Hong 2juan 1, ZHAN G L i 2jun 2, FU D e 2cai
3
(1.E xp erie m ental Center ,H ebei N or m al U niversity ,H ebei S h ij iaz huang 050016,Ch ina ;
2.Colleg e of Che m istry ,H ebei N or m al U niversity ,H ebei S h ij iaz huang 050016,Ch ina ;
3.Colleg e of Che m ical E ng ineering and P har m aceu tics ,H ebei U niversity of S cience and T echnology ,H ebei S h ij iaz huang 050018,Ch ina )
Abstract :Syn thesis of 22(22am ino 242th iazo lyl )222(Z )2m ethoxyi m inoacetic acid 22benzth iazo lyl th i oester ,one of the m o st i m po rtan t in term ediates of the th ird generati on cep halo spo rin s ,w as i m p roved .U sed a m ix tu re of 1,22dich lo rom ethane and to luene in stead of the single 1,22dich lo rom ethane ,the reacti on can be carried ou t successfu lly at room tem p eratu re .P roducts w ith h igh quality w ere ob tained w ithou t fu rther p u 2rificati on and the yield w as over 85p ercen t .T he rep roducib ility of the m ethod in large scale is good .Key words :syn thesis ;22(22am ino 242th iazo lyl )222(Z )2m ethoxyi m inoacetic acid 22benzth iazo lyl th i oester ;an tib i o tic ;cep halo spo rin
收稿日期:2002201228;修回日期:2002202228
作者简介:周鸿娟(1965~),女,高级实验师,主要从事有机化学合成与有机分析工作。
青霉素类和头孢菌素类[1]是临床应用最广、用量最大的一类抗生素,由于它们具有广谱抗菌、过敏反应少、对Β2内酰胺酶具有一定抗菌活性的耐受性等优点,国外对这类药物的研究非常活跃。氨噻肟乙酸分子结构中的氨基噻唑基团增加了抗菌活性,顺式的甲肟基团增强对Β2内酰胺酶的耐受性,它已成为半合成第三代头孢菌素类的重要侧链之一;由它构成的头孢菌素类的品种有十余种[2]。目前主要国产品种有头孢曲松钠(Ceftriaxone Sodium )、头孢噻
肟钠(Cefo tax i m e Sodium )。半合成这类头孢菌类抗
生素的基本途径有(1)采用试剂活化,再与头孢烷酸缩合。(2)采用氨噻肟乙酸苯并噻唑硫酯的合成路线[3]。由于第二条路线具有易于操作流程短等优点,已成为半合成第三代头孢菌素类抗生素的主要生产路线。氨噻肟乙酸苯并噻唑硫酯是半合成这类药物的重要中间体之一。它的品质和收率直接影响到所合成药物的好坏和成本。有关氨噻肟乙酸苯并噻唑硫酯的合成方法,鲜见国内文献报道,国外文
?
746?第12期化 学 世 界
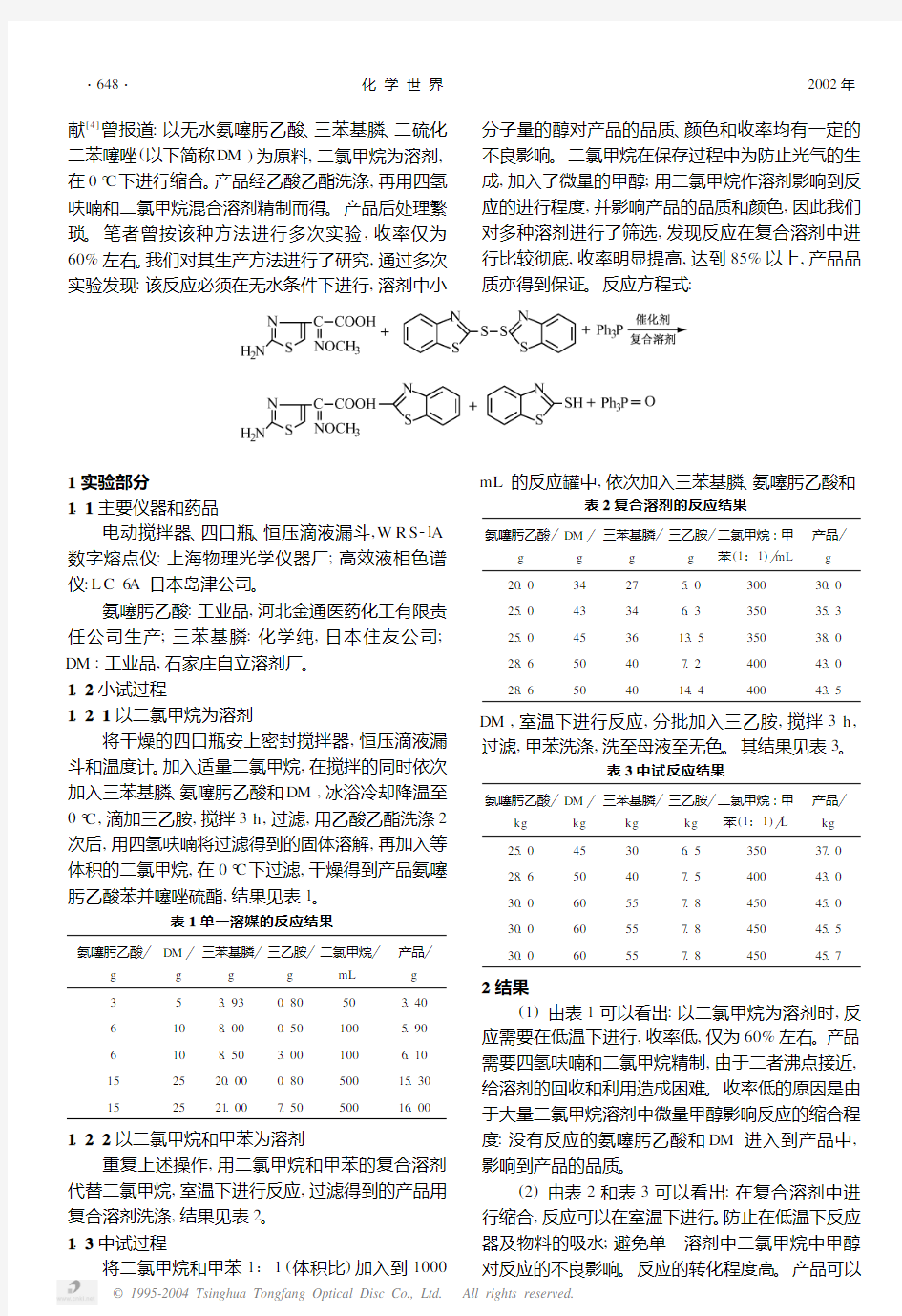
献[4]曾报道:以无水氨噻肟乙酸、三苯基膦、二硫化二苯噻唑(以下简称DM )为原料,二氯甲烷为溶剂,在0°C 下进行缩合。
产品经乙酸乙酯洗涤,再用四氢呋喃和二氯甲烷混合溶剂精制而得。产品后处理繁琐。笔者曾按该种方法进行多次实验,收率仅为60%左右。我们对其生产方法进行了研究,通过多次
实验发现:该反应必须在无水条件下进行,溶剂中小
分子量的醇对产品的品质、颜色和收率均有一定的
不良影响。二氯甲烷在保存过程中为防止光气的生成,加入了微量的甲醇;用二氯甲烷作溶剂影响到反应的进行程度,并影响产品的品质和颜色,因此我们对多种溶剂进行了筛选,发现反应在复合溶剂中进行比较彻底,收率明显提高,达到85%以上,产品品质亦得到保证。反应方程式
:
1 实验部分1.1 主要仪器和药品
电动搅拌器、四口瓶、恒压滴液漏斗,W R S 21A
数字熔点仪:上海物理光学仪器厂;高效液相色谱仪:L C 26A 日本岛津公司。
氨噻肟乙酸:工业品,河北金通医药化工有限责任公司生产;三苯基膦:化学纯,日本住友公司;DM :工业品,石家庄自立溶剂厂。1.2 小试过程
1.2.1 以二氯甲烷为溶剂
将干燥的四口瓶安上密封搅拌器,恒压滴液漏斗和温度计。加入适量二氯甲烷,在搅拌的同时依次加入三苯基膦、氨噻肟乙酸和DM ,冰浴冷却降温至
0°C ,滴加三乙胺,搅拌3h ,过滤,用乙酸乙酯洗涤2次后,用四氢呋喃将过滤得到的固体溶解,再加入等体积的二氯甲烷,在0°C 下过滤,干燥得到产品氨噻肟乙酸苯并噻唑硫酯,结果见表1。
表1 单一溶媒的反应结果
氨噻肟乙酸 g DM g 三苯基膦 g 三乙胺 g 二氯甲烷 mL 产品
g 353.930.80503.406108.000.501005.906108.503.001006.10152520.000.8050015.3015
25
21.00
7.50
500
16.00
1.2.2 以二氯甲烷和甲苯为溶剂
重复上述操作,用二氯甲烷和甲苯的复合溶剂代替二氯甲烷,室温下进行反应,过滤得到的产品用复合溶剂洗涤,结果见表2。1.3 中试过程
将二氯甲烷和甲苯1∶1(体积比)加入到1000
mL 的反应罐中,依次加入三苯基膦、
氨噻肟乙酸和表2 复合溶剂的反应结果
氨噻肟乙酸 g DM g 三苯基膦 g 三乙胺 g 二氯甲烷∶甲
苯(1∶1) mL
产品
g 20.034275.030030.025.043346.335035.325.0453613.535038.028.650407.240043.028.6
50
40
14.4
400
43.5
DM ,室温下进行反应,分批加入三乙胺,搅拌3h ,
过滤,甲苯洗涤,洗至母液至无色。其结果见表3。
表3 中试反应结果
氨噻肟乙酸 kg DM kg 三苯基膦 kg 三乙胺 kg 二氯甲烷∶甲
苯(1∶1) L
产品
kg 25.045306.535037.028.650407.540043.030.060557.845045.030.060557.845045.530.0
60
55
7.8
450
45.7
2 结果
(1)由表1可以看出:以二氯甲烷为溶剂时,反
应需要在低温下进行,收率低,仅为60%左右。产品需要四氢呋喃和二氯甲烷精制,由于二者沸点接近,给溶剂的回收和利用造成困难。收率低的原因是由于大量二氯甲烷溶剂中微量甲醇影响反应的缩合程度:没有反应的氨噻肟乙酸和DM 进入到产品中,影响到产品的品质。
(2)由表2和表3可以看出:在复合溶剂中进行缩合,反应可以在室温下进行。防止在低温下反应器及物料的吸水;避免单一溶剂中二氯甲烷中甲醇对反应的不良影响。反应的转化程度高。产品可以
?846? 化 学 世 界2002年
从复合溶媒直接析出,不夹带没有反应的氨噻肟乙酸和DM,摩尔收率达85%以上,纯度高,不需要精制,可以直接用于下一步合成反应而不影响合成药物的品质。经中试放大,重现性好,收率高,品质稳定,易于工业化。
3 讨论
在氨噻肟乙酸苯并噻唑硫酯的合成过程中,以复合溶媒代替单一溶媒,反应可在室温下进行,反应过程得到明显改善,转化率高;避免没有消耗掉的反应物与产品的共同析出,提高了产品品质,不需精制。中试放大结果证明:重现性好,工业生产过程易于控制。生产的产品为浅黄绿色膨松固体,熔点为133~134°C。经高压液相色谱测定,有效含量达98%以上,与意大利Syneco s r1公司的同类产品水平相当,经国内多个厂家使用,反应良好。
参考文献:
[1] 原正平,王汝龙主编.华工产品手册(药物)[M].北京:
化学工业出版社,1987.145.
[2] 傅德才,赵有贵,等.河北化工[J],1997,(1):52255.
[3] 许思忠.国外医药抗生素分册[J],1995,14(4):272.
[4] Fu rlenm eier A,Hofheinz W,H ub schw erlen C N,et
a l.P reparati on of an ti m icro
b ial12su lfo222oxcazetaid2
inecarboxylic acid derivatives v ia catalytic ester cleav2 age and pharm aceu ticals con tain ing them[P].U S:4
652651,1987203224.
(上接第624页)
超强固体碱K2O Χ2A l2O3。相比之下,该制备方法较常规方法能使固体碱的碱中心原位产生,不会中毒。而Χ2A l2O3载体的存在明显地增强KNO3的分解,表明了KNO3与Χ2A l2O3间存在较强的相互作用。
另外,由于KNO3负载在Χ2A l2O3上常温下并不分解,碱中心只能在活化过程中才产生,因此有效地避免了空气中二氧化碳和水等杂质的毒化,使其在反应中保持着很高的催化效果,具有独特的工业应用价值。
参考文献:
[1] 田部浩三,御园生诚,小野嘉夫,等.新固体酸和碱及
其催化作用[M].北京:化学工业出版社,1989.
[2] 肖晓明.石油化工[J].1994,23(5):3422346.
[3] 李大塘.湘潭师范学院学报[J],1997,18(6):16222.[4] T anabe K,M isono M,O no T,et a l.Stud Su rf Catal
[J].1989,51:1.
[5] W ang Y,Chun Y,Zhu J H,et a l.Ch in J Catal[J],
1999,20(4):4092503.
[6] Chun Y,Zhu J H,Xu Q H.Ch in J Catal[J].1997,
18(4):2982302.
[7] Chun Y Ph.[D]thesis.N an jing:N an jing U n iversi2
ty,1997,109.
[8] 顾庆超,楼书聪,戴庆平,等.化学用表[M].南京:江
苏科学技术出版社,1997,2:51.
[9] X ie Y C,T ang Y Q.A dv Catal[J],1990,37:1.
[10] 沈俭一,章 素,梁东白,等.催化学报[J],1998,9
(2):1152119.
[11] Shen J Y,Guang B,X ia Y F,et a l.Ch in J Ino rg
Chem[J].1995,11(4):4292433.
[12] Zhang S,X ie B Y,Zang J L,et a l.,Ch in J Catal
[J],1980,1(14):2532257.
(上接第627页)
(m o l L)-1?s-1,估算成膜活化能约为117.56kJ m o l,具有典型的化学吸附特征。
4.3 在溶液中形成的TA膜,以化学吸附为主,但表面也存在较多的物理吸附分子。经仔细清洗,可得到较稳定的单层膜,膜质量约为0.377Λg
c m2。
4.4 TA膜对pH值变化敏感,在
5.4~7.6间,膜比较稳定。低pH值,可以破坏膜。pH值在7.6以上,随pH值升高,频移量迅速增加,其主要原因是膜层羧基电离导致膜表面的粘弹性的变化,此时Sauerb rey方程已不适用,需用M artin方程[9]解释。
参考文献:
[1] Cheng Q uan,B rajter2To th A.A nal Chem[J],1995,
67:276722775.
[2] Cheng Q uan,B raiter2To th A.A nal Chem[J],1992,
64:199822000.
[3] L in M ei,L i Q X,R echn itz G A.A nal Ch i m A cta
[J],1999,387:29238.
[4] 聂利华,姚守拙.分析化学[J],1996,24(2):2342241.
[5] 关鲁雄,刘立华.传感器世界[J],2000,6(3):129.
[6] W idrig C A,Chung C,Po rter M D.J E lectroanal
Chem[J],1991,310:3352359.
[7] F ritz M C,H aβhner G,Spencer N D,et a l.L angm u ir
[J],1996,12:607426082.
[8] W ang Juan,F ro stm an L M.W ardM D.J Phys Chem
[J],1992,96:522425228.
[9] M artin S J,F rye G C.A nal Chem[J],1996,63:
227222281.
?
9
4
6
?
第12期化 学 世 界
科研探索 知识创新 与。对羟基苯乙酮在医药、农药、 染料、液晶材料等领域具有重要的应用价值 。 不同生产方法的主要区别在第二步。 方法1:苯酚和乙酐加氯化锌在一定温度下反应,经柱层析可得到对位异构体40%,邻位异构体38%;此方法得率较高, 但反应时间较长,且生成的邻位取代物较多。 方法2:采用三氯化铝——氯化钠复盐作催化剂 合成了对羟 基苯乙酮,收率58.5%纯度98.68%。 综上,我们采用方法3,即以苯酚和乙酐为原料,先进行酯化反应,再通过三氯化铝催化Fries 重排得到产物对羟基苯乙酮,并对酯化反应是否添加催化剂与第二步重排的最佳反应条件进行探究。此方法催化剂易得,产率较高,纯度经精制后很高,是可行的合成方法。3实验 3.1乙酸苯酯的合成 将一定比例的苯酚和已酐混合后加入到50mL 圆底烧瓶中,加入3滴浓硫酸,加热回流一定时间,反应结束后,将反应液冷却至室温,用蒸馏水洗涤至PH 值为6~7,分去水层,保留有机层,用无水硫酸镁干燥后,常压蒸馏,收集190~194℃的馏分,测折光率分析产品。3.2对羟基苯乙酮的合成 在烘干的装有电动搅拌器、温度计、和上部带有干燥管的冷凝管的三口烧瓶中加入一定量的乙酸苯酯和溶剂A ,在剧烈 搅拌下分三次加入一定量的无水三氯化铝,加完后开始加热使反应温度保持在t ℃左右反应一定时间,停止加热。搅拌下加入一定量的水分解多余的无水三氯化铝。将反应液进行水蒸气蒸馏至澄清,将其转移到敞开容器中,冷却至室温后加入 一定量的一定浓度的稀盐酸,至PH 值为1~2。冰盐浴冷却到-2℃析出白色晶体,过滤得对羟基苯乙酮粗品,干燥称重。将粗品转移至小烧杯中加入一定量的水,水浴加热,分去油层后冰盐浴冷却,过滤得白色针状晶体,再次称重,测熔点和红外。 3.3实验结果与讨论 3.3.1反应时间对乙酸苯酯收率的影响 采用酐醇摩尔比1.2,改变反应时间,当回流时间为2h 时, 产率为46.04%,2.5h 时,产率为60.95%,3h 时,产率为67.7%。可见,随着反应时间相对减少,收率逐渐降低。其原因可能是反应时间过短,反应不完全,反应时间过长,逆反应进行程度较大。 3.3.2反应温度对对羟基苯乙酮收率的影响 采用乙酸苯酯、氯苯、催化剂无水三氯化铝摩尔比1:1.2:1.1,改变反应温度,结果表示,随温度升高,对羟基苯乙酮的收率先增加后减少,在70℃时收率最高,大致成抛物线型变化。在相对较低的温度下, 随着温度的升高,单位体积内反应物的活化分子数增多,从而增加了单位时间内单位体积内反应物 分子的有效碰撞的频率,导致反应速率增大
第一章 卤化反应 一、烯丙型、苄基型化合物自由基卤化反应 卤化试剂:NBS 、卤素 溶剂:CCl4、氯仿、苯、石油醚或反应底物自身 二、芳香环上的亲电卤化反应 (一)卤素单质为卤化剂的亲电取代反应 (二)氢卤酸及其盐为卤化剂的卤化反应 (三)胺氮卤化剂为卤化剂的卤代反应(N-氯代丁二酰亚胺NCS ,N-溴代丁二酰亚胺NBS ,N-溴代乙酰胺NBA ,N-氯代乙酰胺NCA ) (四)次卤酸及其衍生物的卤化剂的卤化反应 三、芳香烃卤甲基化反应(Blanc 反应 ) 卤甲基化试剂:甲醛-卤化氢、多聚甲醛-卤化氢、卤甲醚等。 质子酸:硫酸、磷酸、乙酸和Lewis acid 等均可催化反应。 四、不饱和烃与卤素的亲电加成反应 机理: 1桥金属离子历程 2碳正离子历程 OEt O NBS O Br Br CH 3Br hv / Br 2160 ~ 180o C Br Br 85%+Cl 23Cl +HCl MeO CONMe 2MeO CONMe 2Cl TBHP / HCl +MeO CONMe 2 50%35 : 65S H 3C S H 3C X X = Cl 94% Br 83%NXS / solvent 4H 3 CO H 3CO H 3CO Cl Cl t-BuOCl / (C 2H 5)3N-3HF 0o C / CH 2Cl 265%4% ++HCHO +HCl ZnCl 2 Cl
五.不饱和烃与卤化氢、氢卤酸的亲电加成 六、羰基化合物α-位卤化反应 卤化剂:X2、N-卤代酰胺、 次卤酸酯、硫酰卤 溶剂:CCl4, CHCl3, Et2O, AcOH 七、羟基的置换卤化反应 卤化剂:卤化氢、氢卤酸、卤化磷、含硫卤化物 (基本规律:1苄醇=烯丙醇>叔醇>仲醇>伯醇2HI>HBr>HCL>HF) 二章 硝化反应 直接硝化(电子云密度高的芳烃) HO Br HO 2+2Me H H Et Me H H Et Me H H Et Cl Cl Cl OAc Me H H Et AcO Cl + 52% 13% 33%LiCl 69% 8% 21%LiCl CH 3CH=CH 2CH 3CHBrCH 3CH 3CH 2CH 2Br 无过氧化物过氧化物Markovnikov 加成反Markovnikov 加成 O MeOOC HO Br 2O MeOOC HO Br ROH +HX RX +H 2O
D-对羟基苯甘氨酸的制备 制药081(10084349)刘朝阳 1前言 1.1目的 D-对羟基苯甘氨酸是重要的医药中间体,通过查阅国内外有关文献,本文总结了对羟基苯甘氨酸的性质、用途、主要生产路线和生产开发情况。 1.2产品介绍 D-对羟基苯甘氨酸(简称:D-p-HPG)是一种重要的医药精细化学品,主要用于合成β-2-内酰胺类半合成抗菌素,如羟氨苄青霉素(阿莫西林)、头孢克罗、头孢立新、头孢拉定等抗菌药物。这些药物用途广泛,对革兰氏阳性菌、革兰氏阴性菌、弓形体、螺旋体等均有杀灭作用;同时它也用于多种多肽类激素及农药的合成、人工甜味剂的重要中间体。 【结构式】 D-对羟基苯甘氨酸(D-p-hydroxylphenylglycine,D-p-HPG),化学名D-α- 氨基对羟基苯乙酸,分子式(OH)C 6H 4 NH 2 CH 2 COOH,分子量167.2。 【性状】 白色片状结晶,熔点204℃(分解),微溶于乙醇和水,易溶于酸或碱溶液生成盐。 1.3研究意义 D-对羟基苯甘氨酸是一种重要是合成广谱抗生素羟氨卞青霉素和羟基头孢菌素的重要原料,用途广泛。中国是抗生素类药物的生产和需求大国,而且中国制药行业已把半合成青霉素和半合成头孢菌素作为发展重点,因此对D-HPG新工艺的研究具有重要的现实意义。
2合成方法综述 合成方法大致分两类:一类是生物酶催化选择性合成D-HPG,该法选择性高,污染小,但因生物菌培养问题,大规模工业化生产还有一定技术难度;另一类是采用化学方法合成得到外消旋体D,L-对羟基苯甘氨酸(D,L-HPG),再经拆分得到具有光学活性的D-HPG。 2.1D,L-HPG的合成 化学合成是工业上生产D-HPG普遍采用的,但近年来,随着环保要求的不断提高和生物酶技术在手性氨基酸药物中的研究的不断进展,利用生物催化合成 D-HPG逐渐成为研究的热点。 2.1.1生物催化合成法 与化学合成方法相比, 生物催化法具有环境污染小、反应条件温和、选择性和转化率高等优点,但生物菌种的筛选较为困难,投资大,生物酶容易失活,无法大规模连续化生产。因此生物催化合成法仍以实验室研究较多。对于生物催化合成法的研究主要集中在利用D,L-对羟基苯海因(D,L-HPH)为原料经酶催化合 成D-HPG上。 第一步使用D-海因酶作用在底物D,L-HPH上,使其进行不对称开环生成N-氨基甲酰-D-对羟基苯甘氨酸,第二步再将N-氨基甲酰 -D -对羟基苯甘氨酸用化学方法水解脱去氨甲酰基得D-HPG。 该方法的优点在于D-海因酶能选择性水解D-HPH,而L-HPH在碱性条件下可以自发消旋为D,L-HPH,底物的利用率达到100%,但反应第二步采用化学方法水解,污染问题仍较为严重。 2.1.2化学合成法 化学合成因其具有生产工艺简单,易于操作等优点,目前国内外所有文献一致倾向于先合成出外消旋化的D,L-HPG,然后再进行拆分获得D-HPG的两步法。有些方法还包括将不需要的L-HPG进行消旋化。 D,L-HPG的化学合成方法主要有以下几种。 2.1.2.1对甲氧基苯甲醛法 该法是早期用于工业生产D,L-HPG的合成方法。对甲氧基苯甲醛与氰化钠在水溶液或醇溶液中,经环合、加压碱水解和脱甲基,得到D,L-HPG。
乙酸正丁酯的制备 一. 实验内容: 1.以乙酸和正丁醇为原料制备乙酸乙酯。 2.称重并计算产率。 3.完成实验报告和思考题。 二.基本原理: 乙酸和正丁醇的酯化反应为: H+ CH3COOH+CH3CH2CH2CH2OH CH3COOCH2CH2CH2CH3+H2O 为了促使可逆的酯化反应向右进行,可让某一原料过量或连续地移去产物(酯和水)中的一种或全部的方式来达到。 本实验用浓硫酸作催化剂,等量的乙酸与正丁醇反应,采用分水器分水提高乙酸正丁酯的产率。 乙酸正丁酯、正丁醇和水的共沸物组成 组成(质量%) 共沸物沸点/℃ 正丁醇乙酸正丁丁酯水正丁醇-水93.0 55.5 44.5 乙酸正丁酯-水90.7 72.9 27.1 正丁醇-乙酸正丁酯117.6 67.2 32.8 正丁醇-乙酸正丁酯-水90.7 8.0 63.0 29.0 三. 主要仪器、试剂及材料: 1. 主要仪器 圆底烧瓶(50mL)、分水器、球形冷凝管、量筒、分液漏斗、温度计(200℃)、锥形瓶、三角漏斗、直形冷凝管、蒸馏头、尾接管、梨形瓶或圆底烧瓶烧杯(25mL)、台秤、电热套、脱脂棉、阿贝折光仪 2. 试剂及材料 正丁醇11.5mL、冰醋酸7.2mL、浓硫酸0.5mL、10%碳酸钠、无水硫酸镁
四. 实验装置: 五. 实验步骤: 在干燥的50mL圆底烧瓶中,加入11.5mL(0.125mol)正丁醇和7.2mL(0.125mol)冰醋酸,摇匀。再小心地进入3~4滴浓硫酸,并加入2~3粒沸石。按装分水器和球形冷凝管,并在分水器中预先加水至略低于支管口,接通冷却水,用电热套加热回流,把反应一段时间生成的水逐渐分去,保持分水器中水在原来高度,约30~40分钟后不再有水生成,表示反应完毕,停止加热,记录出水量。 冷却后卸下回流冷凝管,在分水器中加水将有机层排入反应瓶中。将反应瓶中的液体倒入分液漏斗中,用10mL水洗涤,分出下层水层,上层有机层用10mL10%碳酸钠溶液洗涤,试验是否仍呈酸性(若仍呈酸性,继续用碳酸钠溶液洗涤直至呈微碱性),分去水层,将酯层用10mL水洗涤至中性。 有机层转入锥形瓶中,加入无水硫酸镁干燥20~30min。 搭好蒸馏装置(使用的玻璃仪器应干燥),小心地将酯液通过漏斗过滤到30mL梨形烧瓶内,加入2~3粒沸石。加热蒸馏,收集124~125℃之间的馏分。 产品称重,计算产率。 用阿贝折光仪测其折光率。
实验七对羟基苯乙酮的合成 一、实验目的 1、掌握Fries重排重排反应的基本原理。 2、熟悉减压蒸馏和水蒸汽蒸馏基本操作。 二、实验原理 醇与酸作用生成酯的反应称作酯化反应。酚类化合物虽然也能起酯化反应, 但比醇困难。这是因为酚中存在的p-π共轭效应,降低了氧周围的电子云密度, 使其亲核性比醇弱。所以酚不能直接与酸成酯,而要与酸酐或酰氯作用才能成酯。 酚酯化在三氯化铝存在下加热,酰基可重排到羟基的邻位或对位,称Fries重排。 通常在低温下易于生成对位异构体。 三、实验材料与设备 1、实验设备与仪器 电动搅拌器、水蒸气发生器、长颈圆底烧瓶、量筒、梨形分液漏斗、蒸馏瓶、直 型冷凝器、球形冷凝器、空气冷凝器、三角烧瓶、尾接管、三口瓶、烧瓶、温度 计。 2、实验材料与试剂 苯酚、醋酐、四氯化碳、硝基苯、氢氧化钠、碳酸氢钠、无水三氯化铝、盐 酸、氢氧化钾、无水硫酸镁、三氯甲烷、无水氯化钙。 四、实验操作步骤 1.乙酰苯酚的制备 取500mL长颈锥形瓶(没有就用500mL三颈瓶替代),加入23.5g苯酚,再加入160ml (10%)NaOH溶液,后加入175g碎冰,最后加入32.5g(30mL)醋酐,猛烈振摇反应容器5min,反应液乳化,生成乙酰苯酚,将混合液倾入500 mL分液漏斗中,加入10 mL CCl 4振摇,静置,分层。水层再用10ml CCl 萃取、合并。酯层用5%~10% NaHCO3 40 mL 4
溶液洗涤至pH7到8,酯层(即CCl 4层)置于250mL三角锥瓶中。用适量无水CaCl 2 约 6克干燥,不时地振摇约1h,然后滤至100 mL蒸馏烧瓶中,旋蒸除去CCl 4 ,然后减压蒸馏收集常压下192~197℃的馏分,称重,计算收率。(油浴开始加热,阶段梯度加热(10+10+5+5+5…℃,当油浴锅指示温度为115℃,温度计指示温度为74℃时开始有馏出液溜出;馏出液呈无色透明。) 2.对羟基苯乙酮的制备(Fries重排反应) 在烘干的装有转子、温度计100mL的三口烧瓶中加入乙酸苯酚10g,硝基苯 25mL,剧烈搅拌下分数次缓慢加入无水三氯化铝16g,加完后开始油浴加热回流 升温至60℃反应2h,停止加热,慢慢倾入到150g冰水中,并迅速搅拌。滴加 6mol/L的盐酸(36%的浓盐酸:水=1:1)酸化至pH1~2,用500mL分液漏斗分出 硝基苯层,用5%~10%KOH溶液中和至微酸性或中性,然后进行水蒸气蒸馏, 至硝基苯蒸净为止(约1h),水层用CHCl 3 提取三次(20mL、15mL、10mL),合并 CHCl 3 提取液置于100mL干燥的三角烧瓶中,加适量的无水硫酸钠干燥,摇匀后 放置约20min,滤除硫酸钠,蒸馏除净CHCl 3 后得粗品。(棕黄色) 3.精制 将对羟基苯乙酮粗品和20 倍量的蒸馏水加入反应瓶中加热至沸腾。分去油 层后添加少量活性炭, 在沸腾状态下脱色 15 m in, 趁热过滤得无色透明液体, 室温下静置, 冷却, 结晶后过滤, 真空干燥得白色针状结晶对羟基苯乙酮。(文 献熔点,对羟基苯乙酮的熔点108~111℃) 五、实验注意事项和该思考的问题 1、(以下为一位同学针对其中一个现象的分析,同学可以思考以下,是否正确) 以硝基苯作溶剂乙酰苯酚在无水三氯化铝的催化作用下进行Fries重排,得 到对羟基苯乙酮;反应液倒入冰水中后,使反应液的温度降低,三氯化铝水解生 成氢氧化铝,溶于水相,对羟基苯乙酮形成其铝盐也溶于水相,这时,反应液分 成三层,上层为水层,中间为絮状的氢氧化铝,下层为硝基苯层,反应液呈酸性, pH为3左右,这是由于三氯化铝水解和对羟基苯乙酮酚羟基上的氢离子电离; 用盐酸酸化至pH1~2,这时,对羟基苯乙酮酚羟基上的铝离子离去而得到氢离子, 不溶于水相而溶于硝基苯;用KOH溶液调pH至中性或微酸性,通过水蒸气蒸馏 出去有机溶剂硝基苯,水层用三氯甲烷(对羟基苯乙酮易溶于三氯甲烷)萃取,
2011年5月第34卷第3期四川师范大学学报(自然科学版) Journal of Sichuan Normal University (Natural Science )May ,2011Vol.34,No.3 收稿日期:2010-09-26 基金项目:四川省青年基金(2010JQ0048)和四川省教育厅自然科学重点项目(09ZA110)基金资助项目 *联系作者简介:王周玉(1977—),女,副教授,主要从事手性药物合成及其方法学、有机小分子催化剂的设计及应用等方面的研究 不活泼芳胺异硫氰酸酯的合成研究 蒋胜前,王周玉* ,蒋珍菊,陈万义,李建惠 (西华大学制药工程系,四川成都610039) 摘要:以不活泼芳胺、二硫化碳(CS 2)、三乙烯二胺(DABCO )、双(三氯甲基)碳酸酯(BTC )为原料合成不活泼芳胺异硫氰酸酯,对合成条件进行了研究,在最优的条件下合成了7个不活泼芳胺异硫氰酸酯,收率高达95%,并利用红外、核磁对其结构进行了表征. 关键词:异硫氰酸酯;不活泼芳胺;三乙烯二胺;双(三氯甲基)碳酸酯中图分类号:O621.3;O643.3 文献标志码:A 文章编号:1001-8395(2011)03-0388-04 doi :10.3969/j.issn.1001-8395.2011.03.023 芳基异硫氰酸酯是一类重要的有机合成中间 体[1-4] ,由于其反应活性高、反应后可引入2个杂原子,在农药、医药、精细化工品领域有广泛的应用.同时,它本身具有防癌、抑癌作用,还可用于测定肽和蛋白质中氨基酸顺序以及作为荧光素标记物[4-9] .目前,市场上销售的芳基异硫氰酸酯品种有限、 价格昂贵,特别是部分不活泼芳胺的异硫氰酸酯.因此,不活泼芳胺异硫氰酸酯的合成具有重要意义. 芳基异硫氰酸酯的合成方法很多 [4,10-26] ,经典 的合成方法是利用胺类物质和硫光气反应,但硫光 气是一种剧毒的挥发性物质,对于一般实验室制备不安全;另一种常用方法是利用胺类物质和二硫化碳反应,再在金属盐或者三氯氧磷的作用下生成异硫氰酸酯,这一方法对于供电子活泼芳胺具有很高的收率,但对吸电子不活泼芳胺收率非常低,部分吸电子芳基胺甚至不反应;对于不活泼芳胺异硫氰酸酯的合成目前较好的方法是采用固体光气法,但是,很少有人对反应条件进行研究.本文对该方法的反应条件进行了研究,并在最优的条件下合成了几个难合成的不活泼芳胺异硫氰酸酯,利用红外和核磁共振对其结构进行了表征. 1实验部分 1.1 主要仪器与试剂傅立叶红外光谱仪(NICO-NET380,天津市拓普仪器有限公司);核磁共振仪 (Brucker -600);WRS -2微机熔点仪(上海申光仪 器仪表有限公司);BP2105型电子天平(北京赛多利斯公司);ZF -2型三用紫外仪(上海安亭电子仪器厂). 实验所用试剂均为市售化学纯或分析纯.1.2 不活泼芳胺异硫氰酸脂的合成不活泼芳胺与CS 2、 DABCO 反应,然后再在BTC 的作用下生成异硫氰酸酯,合成路线如图1所示. 在100mL 的三口瓶中加入芳胺1、 DABCO 和溶剂甲苯,搅拌至全部溶解,慢慢滴加CS 2,室温下搅拌反应12h ,过滤,滤出固体用甲苯洗3次,烘干,得芳基二硫代甲酸盐粉末2粗品.将2悬浮于溶剂中,冷却至10?以下,将BTC 溶于溶剂滴入,室温下反应2h ,再回流5h ,使之完全反应,过滤,滤液减压浓缩,色谱柱层析,得到产品. 1.3红外光谱的测定将样品研碎与KBr 混合压 片, 在红外光谱仪上进行测定,找出相关基团的特征吸收峰. 1.4核磁共振碳谱的测定将样品溶于氘代氯 仿,在核磁共振仪上进行测定. 2结果与讨论 2.1 反应条件对产物收率的影响本文以3,5-二三氟甲基苯胺为模板反应,研究了DABCO 、CS 2、BTC 用量以及第二步溶剂对产物收率的影响,实验结果列于表1.结果表明,在其它条件不变的情况
“乙酸正丁酯的制备”实验报告 班级:工艺一班 实验组号:1-8 同组姓名 实验时间 撰写实验报告时间:2011 年12 月10 日
1 实验目的 (1)初步了解和掌握化工产品开发的研究思路和实验研究方法。 (2)学会组织全流程实验,并获得高纯度的产品。 (3)学会分析实验流程及实验结果,提出实验改进方案。 二、实验原理 酸与醇反应制备酯,是一类典型的可逆反应: 为提高产品收率,一般采用以下措施: 1、使某一反应物过量; 2、在反应中移走某一产物(蒸出产物或水); 3、使用特殊催化剂 用酸与醇直接制备酯,通常有三种方法。 第一种是共沸蒸馏分水法,生成的酯和水以沸臃物的形式蒸出来,冷凝后通过分水器分出水,油层回到反应器中。 第二种是提取酯化法,加入溶剂,使反应物、生成的酯溶于溶剂中,和水层分开。 第三种是直接回流法,一种反应物过量,直接回流。 制备乙酸正丁配用共沸蒸馏分水法较好。为了将反应物中生成的水除去,利用酯、酸和水形成二元或三元恒沸物,采取共沸蒸馏分水法。
使生成的酯和水以共沸物形式逸出,冷凝后通过分水器分出水层,油层则回到反应器中。 三、仪器、试剂与装置 仪器蒸馏装置玻璃磨口仪器、球形冷凝管、分水器、圆底烧瓶(250ml)、温度计(200℃)、锥形瓶(50ml)、烧杯(400ml)、油浴锅、分液漏斗、量筒(10ml、50ml)、电热套、铁架台、铁夹及十字头、铁圈、橡胶水管、天平 试剂正丁醇(23ml,0.25mol)、冰醋酸(16.5ml,0.28mol稍微过量)、KHSO4 1g (催化剂)、NaCl、无水硫酸镁、冰块、沸石、甘油、pH试纸 装置
邻羟基苯乙酮项目技术调查报告 有机0911 朱耀 43 第一章产品及原料介绍 1.1 邻羟基苯乙酮 中文名称:2-羟基苯乙酮;1-(2-羟苯基)-乙酮;邻羟基苯乙酮;邻乙酰基苯酚;英文名称:1-(2-hydroxyphenyl)-Ethanone;o-hydroxy-acetophenon;1-(2-hydroxyphenyl)ethanone;;2'-hydroxy-acetophenon CAS: 118-93-4 ,分子式: C8H8O2 ,分子质量:136.15 ,沸点: 213℃,熔点: 4-6℃,性质描述: 浅绿至黄色油状液体。沸点 213℃/95.6kPa(717mmHg),106℃/2.3kPa(17mmHg),相对密度 1.131,折光率 1.5584,闪点98。 用途: 心律平的中间体。 结构式: 1.2苯酚 相对分子量或原子量94.11,密度1.071,熔点(℃)40.3,沸点(℃)182 ,折射率1.5425(41),毒性LD50(mg/kg) 大鼠经口530。 性状:无色或白色晶体,有特殊气味。在空气中因为被氧化而显粉红色 溶解情况:溶于乙醇、乙醚、氯仿、甘油、二硫化碳等。易溶于有机溶
液,常温下微溶于水,当温度高于65℃时,能跟水以任意比例互溶。 用途:用于制染料合成树脂、塑料、合成纤维和农药、水杨酸等。作外科消毒,消毒能力大小的标准(石炭酸系数)。 制备或来源:由煤焦油经分馏,由苯磺酸经碱熔。由氯苯经水解,由异丙苯经氧化重排。 其他:加热至65℃以上时能溶于水(在室温下,在水中的溶解度是9.3g,当温度高于65℃时能与水混溶),有毒,具有腐蚀性如不慎滴落到皮肤上应马上用酒精(乙醇)清洗,在空气中易被氧化而变粉红色。在民间有土方用石炭酸来治皮肤顽疾,以毒攻毒,如用来治脚底起泡。 1.3乙酐 中文名称:乙酸酐,英文名称:Acetic Anhydride。别名:醋酸酐;醋酐;乙酐;Ac2O 无水醋酸; 分子式:C4H6O3;(CH3CO)2O。外观与性状:无色透明液体,有刺激性气味(类似乙酸),其蒸气为催泪毒气。分子量:102.09 。蒸汽压:1.33kPa/36℃ 闪点:49℃。熔点:-73.1℃。沸点:138.6℃ 溶解性:溶于苯、乙醇、乙醚,氯仿;渐溶于水(变成乙酸)。 密度:相对密度(水=1)1.08;相对密度(空气=1)3.52 。 折光率:n20D 1.450 。稳定性:稳定。 1.4氯苯 中文名称:氯苯、一氯代苯。英文名称:chlorobenzene、monochlorobenzene CAS: 108-90-7 。分子式: C6H5Cl 。分子量: 112.56 。熔点(℃): -45.2 沸点(℃): 132.2 。相对密度(水=1): 1.10 。相对蒸气密度(空气=1): 3.9 饱和蒸气压(kPa): 1.33(20℃) 。临界温度(℃): 359.2 。临界压力(MPa): 4.52 辛醇/水分配系数的对数值: 2.84 。闪点(℃): 28。引燃温度(℃): 590 爆炸上限%(V/V): 9.6。爆炸下限%(V/V): 1.3 。外观与性状:无色透明液体,具有不愉快的苦杏仁味。 溶解性:不溶于水,溶于乙醇、乙醚、氯仿、二硫化碳、苯等多数有机溶剂。主要用途:作为有机合成的重要原料。
二苯基羟基乙酸的合成 摘要用二苯乙二酮作为反应物,以氢氧化钾和乙醇为催化剂,制备二苯基羟基乙酸。产物为白色细晶 体,净重1.56g,产率56.9%;通过氢氧化钠溶液滴定测定产物纯度是100.05%。 关键词二苯基羟基乙酸,多步骤有机反应,混合溶剂重结晶技术,滴定方法 1引言 本实验即应用上回实验的产物二苯乙二酮制备二苯基羟基乙酸。本实验的目的是通过此实验掌握混合溶剂重结晶技术,并了解多步骤有机反应。 2合成原理 二苯乙二酮为α-二酮,与氢氧化钾溶液回流,重排成α-羟基酸盐即二苯乙醇酸钾盐,称为二苯乙醇酸重排。由于反应中形成稳定的羧酸盐,使此重排成为一个不可逆的过程。 二苯乙醇酸也可直接由安息香与碱性溴酸钠溶液一步反应来制备,得到高纯度的产物。 图表 1 制备过程反应式 图表 2 二苯乙醇酸重排机理 3滴定原理 3.1氢氧化钠标准溶液标定原理 本实验产物二苯基羟基乙酸的滴定以氢氧化钠溶液作为标准溶液,而氢氧化钠标准溶液的标定通过邻苯二甲酸氢钾进行。 邻苯二甲酸氢钾()可由邻苯二甲酸酐与氢氧化钾作用而得,分子量为204.22g/mol。常用做滴定分析中的基准物质,用作制备标准碱溶液的基准试剂和测定pH值的缓冲剂,可与氢氧化钠反应生成邻苯二甲酸钾钠。通过邻苯二甲酸氢钾标定的氢氧化钠标准溶液的浓度计算式为: C NaOH(aq)=m邻邻邻邻邻邻邻 204.22×1 V NaOH(aq) 3.2氢氧化钠标准溶液滴定原理 图表 4 酸碱滴定反应式 图表 3 邻苯二甲酸氢钾结构式
产物二苯基羟基乙酸作为酸与氢氧化钠反应式量比为1:1。事先在二苯基羟基乙酸中滴加两至三滴酚酞试剂作为指示剂,当用氢氧化钠标准溶液滴定至恰好显浅粉色且半分钟只内不退色时即为滴定终点。通过氢氧化钠标准溶液滴定二苯基羟基乙酸的质量计算式为: m邻邻邻邻邻邻邻=C邻邻邻邻邻邻邻邻×V邻邻邻邻邻邻邻邻×228.2注意事项:由于从二苯乙醇酸钾盐制备二苯基羟基乙酸的过程用到了盐酸,遗留在二苯基羟基乙酸中的盐酸很可能会导致氢氧化钠溶液滴定得到的结果偏大,纯度甚至超过百分之百;为了得到更为准确的实验结果,洗涤产物时应尽量将产物多清洗几次,测定pH值至洗涤废液pH值接近7为止。 4实验部分 4.1实验条件 实验试剂:二苯乙二酮,乙醇,氢氧化钾,蒸馏水,浓盐酸,刚果红试纸,活性炭,氢氧化钠溶液,邻苯二酸氢钾,酚酞溶液。 实验仪器:圆底烧瓶,茄形瓶,烧杯,磁力搅拌器,油浴装置,球形冷凝管,减压抽气装置,漏斗,花式滤纸,玻璃棒,烘箱,锥形瓶,加料漏斗,布氏漏斗,酸式滴定管。 4.2二苯乙醇酸钾盐的合成 在50 mL 圆底烧瓶中加入二苯乙二酮2.52 g与15 mL 95%乙醇,加热溶解,滴加氢氧化钾2.7 g 溶于5 mL水的溶液,磁力搅拌反应并回流30 min。然后将反应混合物转移到小烧杯中,在冰水浴中放置析出二苯乙醇酸钾盐的晶体。抽滤,并用少量冷乙醇洗涤晶体。 4.3二苯基羟基乙酸的合成 将过滤出的钾盐溶于70 mL水中,滴加2 滴浓盐酸,少量未反应的二苯乙二酮成胶体悬浮物,加入活性炭脱色约两平勺,趁热过滤。滤液冷却至室温,用5%的盐酸酸化至刚果红试纸变蓝,保持搅拌保证产物松散,在冰水浴中冷却使结晶完全。抽滤,用冷水洗涤几次以除去晶体中的无机盐和盐酸。产物在85℃烘箱中干燥至恒重。 4.4滴定过程 4.4.10.1 mo l·L -1NaOH标准溶液的配制与标定 准确称取4.0 g 氢氧化钠溶于1 L蒸馏水中,配制0.1 mo l·L -1的标准溶液。 准确称取0.4 g至0.6 g 邻苯二甲酸氢钾基准物质两份分别于两个250 mL 锥形瓶中,加入40至50 mL水使之溶解,加入3 滴酚酞指示剂,用0.1mo l·L -1氢氧化钠标准溶液滴定至呈微红色,保持半分钟内不退色,即为终点。 4.4.2产品纯度的测定
Chinese Journal of Organic Chemistry NOTE * E-mail: sunxiaoqiang@https://www.doczj.com/doc/ae15737589.html, Received March 18,2015; revised April 16, 2015; published online May 14, 2015. Project supported by the NSFC (No. 21002009), Major Program for the Natural Science Research of Jiangsu Colleges and Universities (Nos. 12KJA150002, 14KJA150002), the Scientific and Technological Project of Jiangsu Province (BY2014037-01), and Qing Lan Project of Jiangsu Province. 国家自然科学基金(No. 21002009), 江苏省高校自然科学研究重大项目(Nos. 12KJA150002, 14KJA150002), 江苏省科技项目(BY2014037-01)和江苏省青蓝工程资助项目. Chin. J. Org. Chem. 2012, 32, xxxx ~xxxx ? 2012 Chinese Chemical Society & SIOC, CAS https://www.doczj.com/doc/ae15737589.html,/ 1 异硫氰酸酯的合成改进 李正义 石 嵩 庄 跃 郑崇谦 张金龙 孙小强* (江苏省绿色催化材料与技术重点实验室 常州大学石油化工学院 常州 213164) 摘要 以硫代氯甲酸苯酯和伯胺为原料, 无需额外加碱条件下分别通过一步或两步反应实现异硫氰酸酯的合成. 一步合成法对高活性胺底物比较有效, 而两步合成法具有广泛的底物适用范围. 该方法具有环境友好、原料易得、低毒和操作简便等许多优点, 具有很好的工业应用前景. 关键词 异硫氰酸酯; 硫代氯甲酸苯酯; 伯胺; 合成 Improvement of the Synthesis of Isothiocyanates Li, Zhengyi Shi, Song Zhuang, Yue Zheng, Chongqian Zhang, Jinlong Sun, Xiaoqiang * (a Jiangsu Key Laboratory of Advanced Catalytic Materials and Technology, School of Petrochemical Engineering ,Changzhou University, Changzhou 213164) Abstract Isothiocyanates were prepared from the reactions of phenyl chlorothionoformate and various primary amines vis one-step or two-step process without additional base. The one-step process is good at highly active amines, while two-step approach is suitable for all kinds of amines. This method has many merits such as environment-friendly, easily available raw materials, low toxicity and convenient operation, which has a good application prospect in industry. Keywords isothiocyanate; phenyl chlorothionoformate; primary amine; synthesis 异硫氰酸酯及其衍生物是一类重要的有机合成中间体,可参与多种有机反应, 用于合成多种含氮和硫的化合物,尤其是杂环骨架的构建[1].许多天然和人工合成的异硫氰酸酯具有很强的生物活性[2], 如抗菌消炎、抗氧化、抗癌、杀虫、除草等活性, 已广泛应用于医药、农药、染料等工业产品的制备[3]. 异硫氰酸酯的人工合成方法种类繁多[4], 其中以伯胺为原料制备异硫氰酸酯是一种相对简便的途径, 主要涉及三种不同方法 (Scheme 1). 第一种方法是硫代光气合成法[5], 由硫代光气与伯胺直接反应生成异硫氰酸酯, 但硫代光气是剧毒的易挥发的液体, 其生产、运输和使用都不安全, 尽管随后发展了许多硫羰基转移试剂[6]如硫代双光气、硫代三光气、硫羰基二咪唑、二(2-吡啶)硫代碳酸盐等, 但这些试剂大多难以获取, 且反应易得到硫脲副产物; 第二种方法是二硫化碳法[7], 由伯胺与二硫化碳在碱的作用下形成二硫代氨基盐, 再与脱硫试剂反应制备异硫氰酸酯, 但这些反应大多条件苛刻, 需要分步进行, 反应时间较长, 后处理繁琐, 且该方法大多只适合于制备烷基和富电子的异硫氰酸酯; 第三种方法是硫代氯甲酸苯酯法, 由Burke 在2000年首次报导[8]. 该方法相对较绿色, 可利用低毒的硫代氯甲酸苯酯替代剧毒的硫光气或二硫化碳, 与伯胺反应生成硫代氨基甲酸酯中间体, 但需要在三氯硅烷和三乙胺共同作用下脱保护生成异硫氰酸酯. 随后本课题组对该方法先后进行了两次改进, 分别以三乙胺[9]或固体氢氧化钠[10]为碱, 硫代氯甲酸苯酯与伯胺在室温下一步或两步反应即可高收率地制备各种类型的异硫氰酸酯衍生物. 有趣
16 1-α-羟基苯乙酸也就是扁桃酸,通常情况下被称之为苦杏仁酸,在其分子结构中由于存在一个手性碳又被称之为手性分子。在制药过程中,1-α-羟基苯乙酸有着加强的使用范围,在治疗血管堵塞疾病中通常对其合成的药物进行临床运用,同时在减肥药物以及抗肿瘤药物中也有着相应的运用。另一方面,1-α-羟基苯乙酸具有较强的分解性能,是当前较为常见的有机酸种类拆分剂,致使其拥有较为良好的发展前景。文章主要对一种完善的化学法进行使用对1-α-羟基苯乙酸进行拆分,也就是将使用钙离子沉淀剂转变为使用镁离子、钙离子沉淀剂,科学有效的对非对应异构体盐进行分解。 1?实验仪器与方法1.1?实验仪器 该实验主要使用型号为WZZ-1的自动指示旋光仪、型号为AB104的电子分析天平、熔点仪、恒温水浴锅等仪器;使用的相关试剂为含量≥95%的1-α-羟基苯乙酸,含量≥95%的盐酸伪麻黄碱、无水乙醇、C 4H 10O、 C 6H 6等。 1.2?试验方法 首先,实验拆分原理。该实验的拆分原理主要是依据?p o Ca d d Ca d d ..2?? 这一化学反应式进行的。其次,拆分工艺流程。研究人员在实际研究实验过程中利用相关设备称取3.8g 1-α-羟基苯乙酸,在将其溶解在20mL的无水乙醇中进行搅拌处理,使两者之间充分的进行融合,称取3.4g的盐酸伪麻黄碱将其与20mL的无水乙醇进行充分融合,通过对其进行搅拌处理提高两者之间的融合度,之后在将两种溶解进行融合并放置在温度为40℃的水溶液 中保温1h左右,对乙醇进行回收,获得相应的胶状物质, 再添加相应的沉淀剂,如含有?? 2Ca n n ?与???Э? 2Mg ??比例为2∶1的 50mLNaClO 3溶液中,使其静止4h,进一步获得颜色为灰白色的固体物质,也就是伪麻黄碱1-α-羟基苯乙酸钙盐的沉淀物质,通过过滤以及抽滤等方法对伪麻黄碱1-α-羟基苯乙酸钙盐进行获取。再次,1-α-羟基苯乙酸钙盐水解。研究人员利用相关设备将钙盐放置在100mL的烧瓶中,添加10mL的蒸馏水,对其进行充分的搅拌,同时结合实际情况添加高东渡氯化氢,对其pH值进行调整使其为1,对其仍进行充分搅拌直至成为固体物质,将其安放在温室中放置10min左右,进行3次乙醚萃取,融合成乙醚液,无水硫化钠干燥,对乙醚进行回收,获得白色的固 体物质1-α-羟基苯乙酸1.7g左右,mp值为119℃指120℃(文献值通常为119℃),光学纯度为99.2%。最后,1-α-羟基苯乙酸纯化处理。研究人员利用设备将1-α-羟基苯乙酸放置在100mL的圆底烧瓶内,再添加15mL的苯,对其进行回流加热至沸腾状态,当固体全部融化溶液成为透明时停止加热,将其安置在温室环境等待结晶现象的发生。在发生结晶现象以及冷却后进行抽滤处理,再使用相应数量的石油醚对结晶体进行洗涤,提高其干燥速度。最终获得白色、重量为1.5g的结晶体,对其旋光度以及熔点等进行检测。 2?实验结论 该实验项目主要是将盐酸伪麻黄碱与1-α-羟基苯乙酸融合形成盐,再通过使用钙离子与镁离子沉淀剂形成d.d-Ca盐沉淀物质,其中DL-盐酸肉碱主要存在于水中,致使1-α-羟基苯乙酸与d-α-羟基苯乙酸进行充分分离。通常升恒的d.d-Ca盐主要为拆分技术中的重要工作项目对拆分效率有着较为直接的影响,因此在实际拆分期间研究人员应对d.d-Ca盐与d.d-Mg盐进行充分的检测。 在对相关图谱进行分析过程中得知,1-α-羟基苯乙酸碳酸根的吸收峰值在达到1617cm -1时,成盐开始消失,进一步导致d.d-Ca盐羧酸根负离子峰值到1647cm -1,盐酸伪麻黄碱—NHR峰值快速消失。另一方面在d.d-Ca盐中铵盐吸收峰值达到2478cm -1时,可证明1-α-羟基苯乙酸碳与盐酸伪麻黄碱形成盐,其中核磁共振图像也可对其进行证明,其中钙离子对已有结构的影响则不能进行相应的显示,致使出现沉淀现象的主要原因还缺乏相应的明了性,需科研人员对其进行深入分析。对镁离子进行添加是1-α-羟基苯乙酸拆分工艺优化的重点,同时在钙离子与镁离子摩尔比达到2∶1时,其拆分效果最为明显。 3?结束语? 综上所述,在对1-α-羟基苯乙酸拆分工艺研究过程中,科研人员通过相应的原理对其进行分析与研究,通过镁离子与钙离子的同时使用进一步对1-α-羟基苯乙酸拆分工艺进行完善。 参考文献? [1]熊正龙,吴桂荣.1-α-羟基苯乙酸拆分工艺研究[J].新疆医科大学学报,2012(1). [2]吴桂荣,杨晓芝.一种由钙离子参与的光学拆分[J].大学化学,2016(5). 1-α-羟基苯乙酸拆分工艺研究 安雪飞 国药集团威奇达药业有限公司 山西 大同 037300 摘要:主要对1-α-羟基苯乙酸拆分工艺进行分析,结合当下1-α-羟基苯乙酸拆分工艺的发展现状,从实验仪器与试剂、实验结果与解析、实验结论等方面进行深入研究与探索,主要目的在于更好地推动1-α-羟基苯乙酸拆分工艺研究的发展与进步。 关键词:1-α-羟基苯乙酸?拆分工艺 沉淀剂 Resolution?process?of?1-α-hydroxy?benzene?acetic?acid? An?Xuefei Sinopharm Weiqida Pharmaceutical Co.,Ltd.,Datong 037300,China Abstract:This?article?describes?the?resolution?processes?of?1-α-hydroxy?benzene?acetic?acid,covering?the?experimental?instruments?and?reagents,experimental?results?and?analysis?as?well?as?experimental?conclusions?on?the?basis?of?the?current?development?status?of?the?processes?to?promote?the?development?of?the?processes. Keywords:1-α-hydroxy?benzene?acetic?acid;resolution?process;precipitation?agent
第41卷第11期 辽 宁 化 工 Vol.41,No. 11 2012年11月 Liaoning Chemical Industry November,2012 收稿日期: 2012-10-04 异硫氰酸烯丙酯合成的研究进展 刘学勇,刘 瑶 (沈阳有色金属研究院, 辽宁 沈阳 110141) 摘 要:异硫氰酸烯丙酯是一种重要的有机合成中间体,其合成及应用受到广泛关注。本文阐述了异硫氰酸烯丙酯的性质及用途,归纳了其主要合成方法及研究现状,并对其进一步研究进行了展望。随着经济的发展,异硫氰酸烯丙酯必定具有更广阔的应用前景。 关 键 词:异硫氰酸烯丙酯; 合成; 应用 中图分类号:TQ 225 文献标识码: A 文章编号: 1004-0935(2012)11-1164-03 异硫氰酸烯丙酯,又称人造芥子油,是一种重要的有机合成中间体,结构式为CH=CH—CH 2—N=C=S,分子量为99.15,它是一种无色至淡黄色透明油状液体,贮藏期间颜色逐渐变深,有较强催泪性,伴有强烈的芥籽似刺激性臭味和辣味,无旋光性[1] 。它是芥子油的有效成分,在医药、染料、矿物加工、食品等方面有广泛应用。它是生产无氰镀铜重要添加剂烯丙基硫脲的一种主要原料, 也用做药物制造、发泡的毒气和擦剂等方面。异硫氰酸烯丙酯在自然界中主要存在于十字花科植物中。目前,人工合成异硫氰酸烯丙酯为构成这一类化合物的主体。其合成方法较多,本文就其合成方法的研究现状进行阐述。 1 异硫氰酸烯丙酯研究进展 目前国内天然生产芥末油工艺主要有两种[2] ,一种是利用蒸馏设备,将芥末籽粉碎、炒拌,静态蒸馏,取其精油,最后再与其它植物油勾兑,提纯制得;另一种是将芥末籽粉碎,经水浸泡发制,在带搅拌装置及冷凝器的不锈钢反应釜中动态水蒸汽蒸馏,馏出物用植物油经萃取,精制后即为异硫氰酸烯丙酯。但从植物中提取产品生产方法能耗大, 收率低, 成本高, 很难以形成规模生产。 1.1 国内外合成异硫氰酸烯丙酯主要方法 (1)氯甲酸丙烯酯与硫氰酸钾化学反应过程; 2-CH=CH 2 + KSCN CH 2=CH-CH 2NCS (30.9%) O (2)丙烯硫醇、丙烯硫醚和氯化氰反应过程, CH 2=CH-CH 2-SH + CNCl CH 2=CH-CH 2NCS CH 2=CH-CH 2-S-CH 2-CH=CH 2 + CNCl CH 2=CH-CH 2NCS (3)丙烯胺与二硫化碳及硫代光气化学反应过 程; CH 2=CH-CH 2 -NH 2 + CSCl 2 CH 2=CH-CH 2NCS CH 2=CH-CH 2 -NH 2 + CS 2 CH 2=CH-CH 2NCS (4)丙烯胺与二硫化碳、三乙胺反应后再与氧氯化磷进行催化反应; CH 2=CH-CH 2 -NH 2 + CS 2 +(C 2H 5)3N CH 2=CH-CH 2NHC-SN(C 2H 5)3S 3 CH 2=CH-CH 2NCS (5)氯丙烯、氰化钠及单质硫磺在有机溶剂介质中进行反应; CH 2=CH-CH 2Cl + NaCN + S CH 2=CH-CH 2NCS (84%) (6)氯丙烯与硫氰酸钠的反应, 溶剂为有机物或水。 CH 2=CH-CH 2 X +MSCN CH 2=CH-CH 2NCS (X=Cl, Br,I; M=k,Na, NH 4) 1.2 国内外有关异硫氰酸烯丙酯合成的研究现状 最近几年, 国内开发了烯丙基异硫氰酸酯的人 工合成工艺[3] , 即常用的硫氰酸盐反应法,通过与烯丙基氯的液固非均相反应, 最后通过常压蒸馏提纯法直接合成异硫氰酸烯丙酯。该法原料易得, 工艺简单, 但反应时间较长, 产品收率低, 后处理较为复杂,并未得到广泛应用。 于跃芹等[4] 以硫氰酸钠和氯丙烯为原料,用乙醇作溶剂, 使硫氰酸盐与烯丙基氯发生均相反应,产