
化学药品注册申报资料要求、申报资料项目
28. 国内外相关的临床试验资料综述。
29. 临床试验计划及研究方案。
30. 临床研究者手册。
31. 知情同意书样稿、伦理委员会批准件。
32. 临床试验报告。
四、申报资料项目表及说明
注:1. “+”指必须报送的资料和/或试验资料。
2?“ 土”指可以用文献资料代替试验资料。
3?“ ―”指可以无需提供的资料。
4?“ *”指按照说明的要求报送资料,如*6,指见说明之第6条。
5?“△”指按照本附件“五、临床试验要求”中第4条执行。
6?文献资料为所申请药物的各项药理毒理(包括药效学、作用机制、一般药理学、毒理学、药代动力学等)研究的文献资料和/或其文献综述资料。
(二)说明
1?申请注册分类1?5的品种,按照《申报资料项目表》的要求报送资料项目1?30 (资料项目6除外);临床试验完成后报送的资料项目包括重新整理的综述资料1?
6、资料项目12和14、临床试验资料28?32以及重新整理的与变更相关的资料和补充的资料,并按申报资料项目顺序排列。
对于注册分类1的品种,临床试验完成后应根据临床期间进行的各项研究的结果,重新整理报送资料项目1?30的全部资料。
同时申请注册属于注册分类3 的原料药和属于注册分类6 的制剂的,其原料药的注册申请应当符合申报生产的要求。
2. 申请注册分类6 的药品,按照《申报资料项目表》的要求报送资料项目1?16 和28?30。需进行临床试验的,在临床试验完成后报送资料项目28?32 以及其他变更和补充的资料,并按申报资料项目顺序排列。
3. 申请注册分类6 的药品,应根据品种的工艺、处方进行全面的质量研究,按国家标准与已上市产品进行质量对比研究。无法按照国家标准与已上市
产品进行质量对比研究的,应按照新药的要求进行质量研究,必要时对国家药品标准项目进行增订和/或修订。
4. 单独申请注册药物制剂,必须提供原料药的合法来源证明文件,一式2 份,分别放入资料项目2的资料(证明性文件)和资料项目13 号的资料(原
料药、辅料的来源及质量标准、检验报告书)中。使用国产原料药的申请人,应当提供该原料药的药品批准证明文件、检验报告书、药品标准、原料药生产企业的营业执照、《药品生产许可证》、《药品生产质量管理规范》认证证书、与该原料药生产企业签订的供货协议、销售发票等的复印件。使用进口原料药的,应当提供与该原料药生产企业或国内合法的销售代理商签订的供货协议、《进口药品注册证》或者《医药产品注册证》、口岸药品检验所检验
报告书、药品标准复印件等。药品注册过程中,研制制剂所用的进口原料药未取得《进口药品注册证》或者《医药产品注册证》的,必须经国家食品药品监督管理局批准。
5. 对用于育龄人群的药物,应当根据其适应症和作用特点等因素报送相应的生殖毒性研究资料。
6. 对于临床预期连续用药6个月以上(含6个月)或治疗慢性复发性疾病而需经常间歇使用的药物,均应提供致癌性试验或文献资料;对于下列情况的药物,需根据其适应症和作用特点等因素报送致癌试验或文献资料:
(1)新药或其代谢产物的结构与已知致癌物质的结构相似的;
(2)在长期毒性试验中发现有细胞毒作用或者对某些脏器、组织细胞生长有异常促进作用的;
(3)致突变试验结果为阳性的。
7. 作用于中枢神经系统的新药,如镇痛药、抑制药、兴奋药以及人体对其化学结构具有依赖性倾向的新药,应当报送药物依赖性试验资料。
8. 属注册分类1 的,一般应在重复给药毒性试验过程中进行毒代动力学研究。
9. 属注册分类1 中“用拆分或合成等方法制得的已知药物中的光学异构体及其制剂” ,应当报送消旋体与单一异构体比较的药效学、药代动力学和毒理学(一般为急性毒性)等反映其立题合理性的研究资料或者相关文献资料。在其消旋体安全范围较小、已有相关资料可能提示单一异构体的非预期毒性(与药理作用无关)明显增加时,还应当根据其临床疗程和剂量、适应症以及用药人群等因素综合考虑,提供与消旋体比较的单一异构体重复给药毒性(一般为3 个月以内)或者其他毒理研究资料(如生殖毒性)。
10. 属注册分类1 中“由已上市销售的多组份药物制备为较少组份的药物” ,如其组份中不含本说明6 所述物质,可以免报资料项目23?25。
11. 属注册分类1 中“新的复方制剂” ,应当报送资料项目22。
12. 属注册分类1 中“新的复方制剂” ,一般应提供与单药比较的重复给药毒性试验资料,如重复给药毒性试验显示其毒性不增加,毒性靶器官也未改变,可不提供资料项目27。
附件2:化学药品注册分类及申报资料要求 一、注册分类 1. 未在国内外上市销售的药品: (1)通过合成或者半合成的方法制得的原料药及其制剂; (2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; (4)由已上市销售的多组份药物制备为较少组份的药物; (5)新的复方制剂; (6)已在国内上市销售的制剂增加国内外均未批准的新适应症。 2. 改变给药途径且尚未在国内外上市销售的制剂。 3. 已在国外上市销售但尚未在国内上市销售的药品: (1)已在国外上市销售的制剂及其原料药,和/或改变该制剂的剂型,但不改变给药途径的制剂; (2)已在国外上市销售的复方制剂,和/或改变该制剂的剂型,但不改变给药途径的制剂; (3)改变给药途径并已在国外上市销售的制剂; (4)国内上市销售的制剂增加已在国外批准的新适应症。 4. 改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原 料药及其制剂。 5. 改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6. 已有国家药品标准的原料药或者制剂。 二、申报资料项目 (一)综述资料 1. 药品名称。 2. 证明性文件。 3. 立题目的与依据。 4. 对主要研究结果的总结及评价。 5. 药品说明书、起草说明及相关参考文献。 6. 包装、标签设计样稿。 (二)药学研究资料 7. 药学研究资料综述。 8. 原料药生产工艺的研究资料及文献资料;制剂处方及工艺的研究资料及文献资料。 9. 确证化学结构或者组份的试验资料及文献资料。 10. 质量研究工作的试验资料及文献资料。 11. 药品标准及起草说明,并提供标准品或者对照品。 12. 样品的检验报告书。 13. 原料药、辅料的来源及质量标准、检验报告书。 14. 药物稳定性研究的试验资料及文献资料。 15. 直接接触药品的包装材料和容器的选择依据及质量标准。 (三)药理毒理研究资料 16. 药理毒理研究资料综述。 17. 主要药效学试验资料及文献资料。
附件 化学药品新注册分类申报资料要求(试行) 第一部分 注册分类1、2、3、5.1类申报资料要求(试行) 一、申报资料项目 (一)概要 1.药品名称。 2.证明性文件。 2.1注册分类1、2、3类证明性文件 2.2注册分类5.1类证明性文件 3.立题目的与依据。 4.自评估报告。 5.上市许可人信息。 6.原研药品信息。 7.药品说明书、起草说明及相关参考文献。 8. 包装、标签设计样稿。 (二)主要研究信息汇总表 9. 药学研究信息汇总表。
10. 非临床研究信息汇总表。 11. 临床研究信息汇总表。 (三)药学研究资料 12. (3.2.S)原料药(注:括号内为CTD格式的编号,以下同)。 12.1(3.2.S.1)基本信息 12.2(3.2.S.2 )生产信息 12.3(3.2.S.3 )特性鉴定 12.4(3.2.S.4)原料药的质量控制 12.5(3.2.S.5)对照品 12.6(3.2.S.6)包装材料和容器 12.7(3.2.S.7)稳定性 13. (3.2.P)制剂。 13.1(3.2.P.1)剂型及产品组成 13.2(3.2.P.2)产品开发 13.3(3.2.P.3)生产 13.4(3.2.P.4)原辅料的控制 13.5(3.2.P.5)制剂的质量控制 13.6(3.2.P.6)对照品 13.7(3.2.P.7)稳定性 (四)非临床研究资料 14.非临床研究资料综述。 15.主要药效学试验资料及文献资料。
16.安全药理学的试验资料及文献资料。 17.单次给药毒性试验资料及文献资料。 18.重复给药毒性试验资料及文献资料。 19.遗传毒性试验资料及文献资料。 20.生殖毒性试验资料及文献资料。 21.致癌试验资料及文献资料。 22.依赖性试验资料及文献资料。 23.过敏性(局部、全身和光敏毒性)、溶血性和局部(血管、皮肤、粘膜、肌肉等)刺激性等特殊安全性试验资料及文献资料。 24.其他安全性试验资料及文献资料。 25.非临床药代动力学试验资料及文献资料。 26.复方制剂中多种成分药效、毒性、药代动力学相互影响的试验资料及文献资料。 (五)临床试验资料 27.临床试验综述资料。 28.临床试验计划及研究方案。 29. 数据管理计划、统计分析计划。 30.临床研究者手册。 31.知情同意书样稿、伦理委员会批准件;科学委员会审查报告。 32.临床试验报告。 33.临床试验数据库电子文件(原始数据库、衍生的分析数据库及其变量说明文件)。
许可事项化学药品注册分类 一、化学药品注册分类 1、未在国内外上市销售的药品: (1)通过合成或者半合成的方法制得的原料药及其制剂; (2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; (4)由已上市销售的多组份药物制备为较少组份的药物;(5)新的复方制剂。 2、改变给药途径且尚未在国内外上市销售的制剂。 3、已在国外上市销售但尚未在国内上市销售的药品: (1)已在国外上市销售的原料药及其制剂; (2)已在国外上市销售的复方制剂; (3)改变给药途径并已在国外上市销售的制剂。 4、改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂。 5、改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6、已有国家药品标准的原料药或者制剂。 二、申报资料项目 (一)综述资料 1、药品名称。 2、证明性文件。 3、立题目的与依据。 4、对主要研究结果的总结及评价。 5、药品说明书样稿、起草说明及最新参考文献。 6、包装、标签设计样稿。
(二)药学研究资料 7、药学研究资料综述。 8、原料药生产工艺的研究资料及文献资料;制剂处方及工艺的研究资料及文献资料。 9、确证化学结构或者组份的试验资料及文献资料。 10、质量研究工作的试验资料及文献资料。 11、药品标准草案及起草说明,并提供标准品或者对照品。 12、样品的检验报告书。 13、辅料的来源及质量标准。 14、药物稳定性研究的试验资料及文献资料。 15、直接接触药品的包装材料和容器的选择依据及质量标准。 (三)药理毒理研究资料 16、药理毒理研究资料综述。 17、主要药效学试验资料及文献资料。 18、一般药理研究的试验资料及文献资料。 19、急性毒性试验资料及文献资料。 20、长期毒性试验资料及文献资料。 21、过敏性(局部、全身和光敏毒性)、溶血性和局部(血管、皮肤、粘膜、肌肉等)刺激性等主要与局部、全身给药相关的特殊安全性试验研究和文献资料。 22、复方制剂中多种成份药效、毒性、药代动力学相互影响的试验资料及文献资料。 23、致突变试验资料及文献资料。 24、生殖毒性试验资料及文献资料。 25、致癌试验资料及文献资料。
化学试剂的纯度分类及标准
————————————————————————————————作者:————————————————————————————————日期: ?
化学试剂的纯度分类及标准 国标试剂:该类试剂为我国国家标准所规定,适用于检验、鉴定、检测 基准试剂(JZ,绿标签):作为基准物质,标定标准溶液。 优级纯(GR,绿标签)(一级品):主成分含量很高、纯度很高,适用于精确分析和研究工作,有的可作为基准物质。 分析纯(AR,红标签)(二级品):主成分含量很高、纯度较高,干扰杂质很低,适用于工业分析及化学实验。 化学纯(CP,蓝标签)(三级品):主成分含量高、纯度较高,存在干扰杂质,适用于化学实验和合成制备。 实验纯(LR,黄标签):主成分含量高,纯度较差,杂质含量不做选择,只适用于一般化学实验和合成制备。 教学试剂():可以满足学生教学目的,不至于造成化学反应现象偏差的一类试剂。 指定级(ZD),该类试剂是按照用户要求的质量控制指标,为特定用户订做的化学试剂。 高纯试剂(EP):包括超纯、特纯、高纯、光谱纯,配制标准溶液。此类试剂质量注重的是:在特定方法分析过程中可 能引起分析结果偏差,对成分分析或含量分析干扰的杂质含量,但对主含量不做很高要求。 色谱纯(GC):气相色谱分析专用。质量指标注重干扰气相色谱峰的杂质。主成分含量高。 色谱纯(LC):液相色谱分析标准物质。质量指标注重干扰液相色谱峰的杂质。主成分含量高 指示剂(ID):配制指示溶液用。质量指标为变色范围和变色敏感程度。可替代CP,也适用于有机合成用。 生化试剂(BR):配制生物化学检验试液和生化合成。质量指标注重生物活性杂质。可替代指示剂,可用于有机合成 生物染色剂(BS):配制微生物标本染色液。质量指标注重生物活性杂质。可替代指示剂,可用于有机合成 光谱纯(SP):用于光谱分析。分别适用于分光光度计标准品、原子吸收光谱标准品、原子发射光谱标准品 电子纯(MOS):适用于电子产品生产中,电性杂质含量极低。 当量试剂(3N、4N、5N):主成分含量分别为99.9%、99.99%、99.999%以上。 电泳试剂:质量指标注重电性杂质含量控制。 此外,还有特种试剂,生产量极小,几乎是按需定产,此类试剂其数量和质量一般为用户所指定。 试剂 reagent 又称化学试剂或试药。主要是实现化学反应、分析化验、研究试验、教学实验、化学配方使用的纯净化学品。 一般按用途分为通用试剂、高纯试剂、分析试剂、仪器分析试剂、临床诊断试剂、生化试
化学药注册分类大变动,CFDA发布《化学药品注册分类改革工作方案》和《化学仿制药生物等效性试验备案管理规定》征求意见稿为贯彻落实国务院《关于改革药品医疗器械审评审批制度的意见》政策要求,根据全国人大常委会《关于授权国务院在部分地区开展药品上市许可持有人制度试点和有关问题的决定》,昨日(11月6日),国家食药监总局官网发布了《化学药品注册分类改革工作方案(征求意见稿)》和《化学仿制药生物等效性试验备案管理规定(征求意见稿)》,面向社会公开征求意见。 《化学药品注册分类改革工作方案(征求意见稿)》 为贯彻落实国务院《关于改革药品医疗器械审评审批制度的意见》政策要求,根据全国人大常委会《关于授权国务院在部分地区开展药品上市许可持有人制度试点和有关问题的决定》,制定本工作方案。 一、工作原则 按照分类科学、标准严格、质量提高的原则,在原有化学药品注册分类的基础上,结合国务院改革意见中有关药品分类的调整原则,对原有化学药品注册分类进行调整和完善。首先,根据药品的安全风险程度,将药品分为新药和仿制药两大类;其次,根据药品原创性和新颖性的不同,将新药进一步分为创新药和改良型新药;第三,在仿制药中,根据被仿制药上市情况不同,进一步细分为对境外上市、境内未上市药品的仿制,对境内上市药品的仿制以及境外上市药品申请境内上市三类。 二、化学药品新注册分类及说明
新药是指未在中国境内外上市销售的药品,将境外上市境内未上市药品纳入仿制药。调整后,化学药品新注册分类共分为1-5类(表1),具体如下: (一)根据物质基础的原创性和新颖性不同,将新药分为创新药(注册分类1)和改良型新药(注册分类2)两类。其中,创新药是指含有新的结构明确的具有生理或药理作用的分子或离子,且具有临床价值的原料药及其制剂,包括用拆分或者合成等方法制得的已知活性成份的光学异构体及其制剂,但不包括对已知活性成份成酯、成盐(包括含有氢键或配位键的盐),或形成其他非共价键衍生物(如络合物、螯合物或包合物),或其结晶水、结晶溶剂、晶型的改变等。 改良型新药是在已知活性成份的基础上,对其结构、剂型、给药途径、适应症、用法用量、规格等进行优化,且具有明显临床优势的药品。结构优化是指对已知活性成份成酯、成盐(包括含有氢键或配位键的盐),或形成其他非共价键衍生物(如络合物、螯合物或包合物),或其结晶水、结晶溶剂、晶型的改变等。 (二)被仿制的参比制剂来源不同,其上市情况存在差异,研制者和监管部门对其上市基础的认识也随之不同,为便于申报,将仿制药分为3-5类。其中,注册分类3是指仿境外上市、境内未上市药品;注册分类4是指仿制境内上市药品;注册分类5是指境外上市的药品申请在境内上市。 仿制药的基本要求是与参比制剂质量和疗效一致,参比制剂须为原研或国际公认的药品。原研药品指境外或境内首先批准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品。国际公认的药品是指与原研药品质量和疗效一致的药品。 表1化学药品新注册分类、说明及包含的情形
附件2: 化学药品注册分类及申报资料要求 一、注册分类 1.未在国内外上市销售的药品: (1)通过合成或者半合成的方法制得的原料药及其制剂; (2)天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3)用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; (4)由已上市销售的多组份药物制备为较少组份的药物; (5)新的复方制剂; (6)已在国内上市销售的制剂增加国内外均未批准的新适应症。 2.改变给药途径且尚未在国内外上市销售的制剂。 3.已在国外上市销售但尚未在国内上市销售的药品: (1)已在国外上市销售的制剂及其原料药,和/或改变该制剂的剂型,但不改变给药途径的制剂; (2)已在国外上市销售的复方制剂,和/或改变该制剂的剂型,但不改变给药途径的制剂; (3)改变给药途径并已在国外上市销售的制剂; (4)国内上市销售的制剂增加已在国外批准的新适应症。 4.改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂。 5.改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6.已有国家药品标准的原料药或者制剂。 二、申报资料项目 (一)综述资料 1.药品名称。 2.证明性文件。 3.立题目的与依据。 4.对主要研究结果的总结及评价。 5.药品说明书、起草说明及相关参考文献。 6.包装、标签设计样稿。 (二)药学研究资料 7.药学研究资料综述。 8.原料药生产工艺的研究资料及文献资料;制剂处方及工艺的研究资料及文献资料。 9.确证化学结构或者组份的试验资料及文献资料。 10.质量研究工作的试验资料及文献资料。 11.药品标准及起草说明,并提供标准品或者对照品。 12.样品的检验报告书。 13.原料药、辅料的来源及质量标准、检验报告书。 14.药物稳定性研究的试验资料及文献资料。 15.直接接触药品的包装材料和容器的选择依据及质量标准。 (三)药理毒理研究资料 16.药理毒理研究资料综述。
附件1 化学药品注册申报资料指南(试行)(第一部分注册分类1、2、3、5.1类) 可编辑范本
目录 一、适用范围 (3) 二、基本要求 (3) (一)申请表的整理 (3) (二)申报资料的整理 (4) 三、申请表 (8) (一)《药品注册申请表》 (8) (二)《小型微型企业收费优惠申请表》 (14) 四、申报资料 (15) (一)申报资料项目 (15) (二)申报资料要求 (18) 五、其他 (24) 附件:1.申报资料袋封面格式 (25) 2.申报资料项目封面格式 (27) 3.申报资料项目目录 (28) 4.化学药品1、2、3、5.1类注册申报资料自查表 (29) 可编辑范本
化学药品注册申报资料指南(试行) 第一部分注册分类1、2、3、5.1类 一、适用范围 化学药品注册分类1、2、5.1类临床试验/新药生产(含新药证书)/上市申请;化学药品注册分类3类仿制药申请。 二、基本要求 (一)申请表的整理 1.种类与份数要求 药品注册申请表、申报资料情况自查表、小型微型企业收费优惠申请表(如适用)各四份,一份为原件;药品研制情况申报表(如适用)、药品注册生产现场检查申请表(如适用)各四份,三份为原件。 2.依据《关于启用新版药品注册申请表报盘程序的公告》,申请表的填报须采用国家食品药品监督管理总局统一发布的填报软件,提交由新版《药品注册申请表报盘程序》生成的电子及纸质文件。(确认所用版本为最新版[以最新发布的公告为准],所生成的电子文件的格式应为RVT文件。各页的数据核对码必须一致,并须与提交的电子申请表一致,申请表及自查表各页边缘应加盖所有申请人或注册代理机构骑缝章。) 3.填写应当准确、完整、规范,不得手写或涂改,并应符合可编辑范本
(一) 化学试剂 1. 化学试剂的分类 化学试剂数量繁多,种类复杂,通常根据用途分为一般试剂、基础试剂、高纯试剂、色谱试剂、生化试剂、光谱纯试剂和指示剂等。采用的标准为国家标准(标以“GB”字样)和行业标准(标以“HG”字样)。食品检验常用的试剂主要有一般试剂、基础试剂、高纯试剂和专用试剂等。 化学试剂的分级: 除此之外还有许多特殊规格试剂,如基准试剂、色谱纯试剂、光谱纯试剂、电子纯试剂、生化试剂和生物染色剂等。使用者要根据试剂中所含杂质对检测有无影响选用合适的试剂。 (1) 一般试剂 根据GB 15346-1994《化学试剂的包装及标志》规定,一般试剂分为三个等级,即优级纯、分析纯和化学纯。通常也将实验试剂列入一般试剂。 (2) 基础试剂 可用作基准物质的试剂叫做基准试剂,也可称为标准试剂。基础准试剂可用来直接配制标准溶液,用来校正或标定其他化学试剂。 如在配置标准溶液时用于标定标准溶液用的基准物 (3) 高纯试剂 高纯试剂不是指试剂的主体含量,而是指试剂的某些杂质的
含量而言。高纯试剂等级表达方式有数种,其中之一是以内处“9”表示,如用于9.99%,99.999%等表示。“9”的数目越多表示纯度越高,这种纯度的是由100%减去杂质的质量百分数计算出来的。 (4) 专用试剂 专用试剂是指具有专门用途的试剂。例如仪器分析专用试剂中色谱分析标准试剂、气相色谱载体及固定液、薄层分析试剂等。与高纯试剂相似之处是,专用试剂不仅主体含量较高,而且杂质含量很低。它与高纯试剂的区别是,在特定的用途中有干扰的杂质成分只须控制在不致产生明显干扰的限度 以下。 表1 .1 化学试剂等级对照表 其他级别化学试剂等级对照表
化学药品注册分类对比(仅供参考) 2016年3月4日,食品药品监管总局发布了《化学药品注册分类改革工作方案》。现将《化学药品注册分类改革工作方案》与《药品注册管理办法》(2007版)相比,有以下这些不同之处:
相较于2015年11月6日《化学药品注册分类改革工作方案(征求意见稿)》中提到的2.5含有已知活性成分的新用法用量和新规格的制剂,在此方案中并未体现,可能只能走补充申请了。 相关注册管理要求 (一)对新药的审评审批,在物质基础原创性和新颖性基础上,强调临床价值的要求,其中改良型新药要求比改良前具有明显的临床优势。对仿制药的审评审批,强调与原研药品质量和疗效的一致。
(二)新注册分类1、2类别药品,按照《药品注册管理办法》中新药的程序申报;新注册分类3、4类别药品,按照《药品注册管理办法》中仿制药的程序申报;新注册分类5类别药品,按照《药品注册管理办法》中进口药品的程序申报。 新注册分类2类别的药品,同时符合多个情形要求的,须在申请表中一并予以列明。 (三)监测期有变动; (四)可以继续按照原规定进行审评审批,也可以申请按照新注册分类进行审评审批。如申请按照新注册分类进行审评审批,补交相关费用后,不再补交技术资料,国家食品药品监督管理总局药品审评中心要设立绿色通道,加快审评审批。符合要求的,批准上市;不符合要求的,不再要求补充资料,直接不予批准。 (五)新注册分类的注册申请所核发的药品批准文号(进口药品注册证/医药产品注册证)效力与原注册分类的注册申请核发的药品批准文号(进口药品注册证/医药产品注册证)效力等同。 (六)国家食品药品监督管理总局组织相关部门细化工作要求,做好受理、核查检查、技术审评及制定、修订相关国家药品标准等工作。 (七)《药品注册管理办法》与本方案不一致的,按照本方案要求执行。
化学试剂常用分类方法有哪些? 试剂分类的方法较多。如按状态可分为固体试剂、液体试剂。按用途可分为通用试剂、专用试剂。按类别可分为无机试剂、有机试剂。按性能可分为危险试剂、非危险试剂等。 化学试剂又叫化学药品,简称试剂。化学试剂是指具有一定纯度标准的各种单质和化合物(也可以是混合物)。要进行任何实验都离不了试剂,试剂不仅有各种状态,而且不同的试剂其性能差异很大。 有的常温非常安定、有的通常就很活泼,有的受高温也不变质、有的却易燃易爆:有的香气浓烈,有的则剧毒……。只有对化学试剂的有关知识深入了解,才能安全、顺利进行各项实验。既可保证达到预期实验目的,又可消除对环境的污染。因此,首先要知道试剂的分类情况。然后掌握各类试剂的存放和使用。 化学试剂的分类 从试剂的贮存和使用角度常按类别和性能2种方法对试剂进行分类。 无机试剂和有机试剂 这种分类方法与化学的物质分类一致,既便于识别、记忆,又便于贮存、取用。 无机试剂按单质、氧化物、碱、酸、盐分出大类后,再考虑性质进行分类。 有机试剂则按烃类、烃的衍生物、糖类蛋白质、高分子化合物、指示剂等进行分类。 危险试剂和非危险试剂 这种分类既注意到实用性,更考虑到试剂的特征性质。因此,既便于安全存放,也便于实验工作者在使用时遵守安全操作规则。 1.危险试剂的分类 根据危险试剂的性质和贮存要求又分为: (1)易燃试剂 这类试剂指在空气中能够自燃或遇其它物质容易引起燃烧的化学物质。由于存在状态或引起燃烧的原因不同常可分为: ①易自燃试剂:如黄磷等。 ②遇水燃烧试剂:如钾、钠、碳化钙等。 ③易燃液体试剂:如苯、汽油、乙醚等。 ④易燃固体试剂,如硫、红磷、铝粉等。 (2)易爆试剂 指受外力作用发生剧烈化学反应而引起燃烧爆炸同时能放出大量有害气体的化学物质。如氯
一、注册分类 1、未在国内外上市销售的药品: (1 )通过合成或者半合成的方法制得的原料药及其制剂; (2 )天然物质中提取或者通过发酵提取的新的有效单体及其制剂; (3 )用拆分或者合成等方法制得的已知药物中的光学异构体及其制剂; ( 4 )由已上市销售的多组份药物制备为较少组份的药物;( 5 )新的复方制剂。 2、改变给药途径且尚未在国内外上市销售的制剂。 3、已在国外上市销售但尚未在国内上市销售的药品: (1 )已在国外上市销售的原料药及其制剂; (2 )已在国外上市销售的复方制剂; (3 )改变给药途径并已在国外上市销售的制剂。 4 、改变已上市销售盐类药物的酸根、碱基(或者金属元素),但不改变其药理作用的原料药及其制剂。 5、改变国内已上市销售药品的剂型,但不改变给药途径的制剂。 6、已有国家药品标准的原料药或者制剂。 二、申报资料项目 (一)综述资料 1、药品名称。 2、证明性文件。 3、立题目的与依据。 4、对主要研究结果的总结及评价。 5、药品说明书样稿、起草说明及最新参考文献。 6、包装、标签设计样稿。
(二)药学研究资料 7、药学研究资料综述。 8、原料药生产工艺的研究资料及文献资料;制剂处方及工艺的研究资料及文献资料。 9、确证化学结构或者组份的试验资料及文献资料。 10、质量研究工作的试验资料及文献资料。 11、药品标准草案及起草说明,并提供标准品或者对照品。 12、样品的检验报告书。 13、辅料的来源及质量标准。 14、药物稳定性研究的试验资料及文献资料。 15 、直接接触药品的包装材料和容器的选择依据及质量标准。 (三)药理毒理研究资料 16、药理毒理研究资料综述。 17、主要药效学试验资料及文献资料。 18、一般药理研究的试验资料及文献资料。 19、急性毒性试验资料及文献资料。 20、长期毒性试验资料及文献资料。 21 、过敏性(局部、全身和光敏毒性)、溶血性和局部(血管、皮肤、粘膜、肌肉等)刺激性等主要与局部、全身给药相关的特殊安全性试验研究和文献资料。 22、复方制剂中多种成份药效、毒性、药代动力学相互影响的试验资料及文献资料。 23、致突变试验资料及文献资料。 24、生殖毒性试验资料及文献资料。 25、致癌试验资料及文献资料。 26、依赖性试验资料及文献资料。 27、动物药代动力学试验资料及文献资料。
中国药品注册的分类说明: 化学药品新注册分类共分为5个类别,具体如下: 1类:境内外均未上市的创新药。指含有新的结构明确的、具有药理作用的化合物,且具有临床价值的药品。 2类:境内外均未上市的改良型新药。指在已知活性成份的基础上,对其结构、剂型、处方工艺、给药途径、适应症等进行优化,且具有明显临床优势的药品。 3类:境内申请人仿制境外上市但境内未上市原研药品的药品。该类药品应与原研药品的质量和疗效一致。原研药品指境内外首个获准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品。 4类:境内申请人仿制已在境内上市原研药品的药品。该类药品应与原研药品的质量和疗效一致。 5类:境外上市的药品申请在境内上市。 涉及到本次双方的合作项目的进口注册类型为5.2类别:境外上市的非原研药品(包括原料药及其制剂)申请在境内上市。 The classification instruction of drug registration in China There are 5 categories of chemical drug new registrations. They are as follows. Category 1: Innovative drugs that are not marketed both domestically and abroad. These drugs contain new compounds with clear structures and pharmacological effects and they have clinical value. Category 2: Modified new drugs that are not marketed both domestically or abroad. With known active components, the drug’s structure, phase, prescription manufacturing process, administration route and indication are optimized and it has obvious clinical advantage. Category 3: The drugs that are imitated by domestic applicants to original drugs that have been marketed abroad but not domestically.This kind of drugs are supposed to have the same quality and effects with original drugs.Original drugs are the foremost drugs that are approved to be marketed domestically and /or abroad with complete and full safety and validity data as marketing evidence. Category 4: The drugs that are imitated by domestic applicants to original drugs that have been marketed domestically. This kind of drugs are supposed to have the same quality and effects with original drugs. Category 5: The drugs that have been marketed abroad are applied to be marketed domestically. The category of the imported registration involved in our collaboration program is category 5.2: non-original drugs( including API and its preparation) that have been marketed abroad are applied to be marketed domestically.
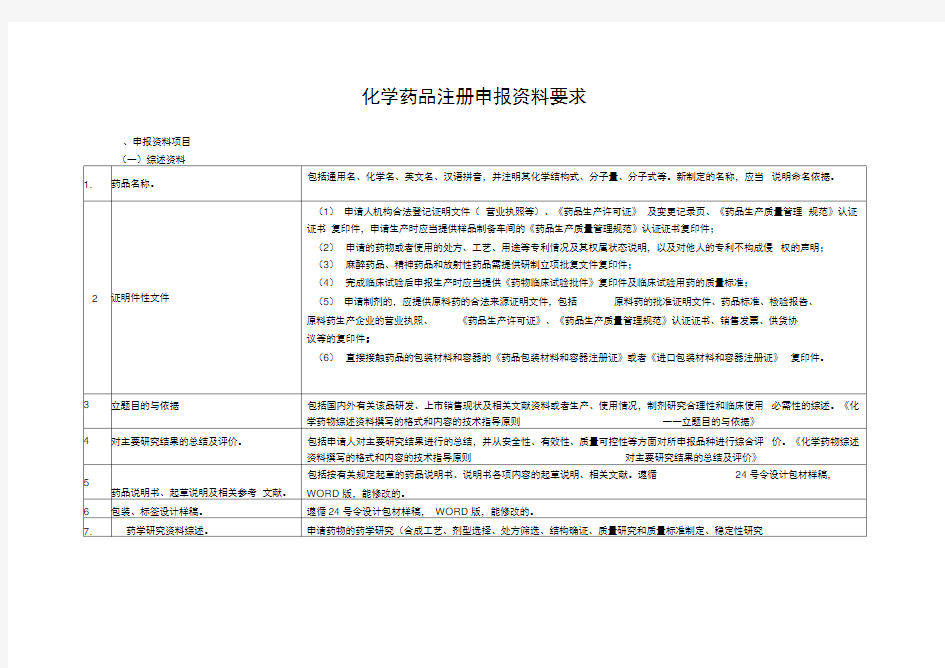
药品注册资料的的准备 一、1~32号申报资料审查的要点和体会 二、全套资料的综合体会 回顾: CTD格式申报资料和附件2格式的比较 2007年《药品注册管理办法》附件2 化学药物申报格式项目(仿制药6类) (一)综述资料 1.药品名称。 2.证明性文件。 3.立题目的和依据。 4.对主要研究结果的总结及评价。 5.药品说明书、起草说明及相关参考文献。 6.包装、标签设计样稿。 (二)药学研究资料 7.药学研究资料综述。 8.原料药生产工艺的研究资料及文献资料;制剂处方及工艺的研究资料及文献资料。 9.确证化学结构或者组份的试验资料及文献资料。 10.质量研究工作的试验资料及文献资料。 11.药品标准及起草说明,并提供标准品或者对照品。 12.样品的检验报告书。 13.原料药、辅料的来源及质量标准、检验报告书。 14.药物稳定性研究的试验资料及文献资料。 15.直接接触药品的包装材料和容器的选择依据及质量标准。 (三)药理毒理研究资料 16.药理毒理研究资料综述。 (四)临床试验资料 28.国内外相关的临床试验资料综述。 CTD格式的申报资料仅替代(二)药学研究资料部分,其余内容保持不变。 一、1~32号申报资料形式审查要点和体会 为便于梳理和阐述,对同一号资料分为以下三种情形: A:新药报临床 B:新药报生产(A、B:包括注册分类1~5类) C:仿制药(注册分类6) 1号资料(药品名称): A:包括通用名、商品名、化学名、英文名、汉语拼音,并注明其化学结构式、分子量、分子式等(见相关要求)。 如果是新剂型、新命名、应附上药典委员会命名的复函 补充:对于缓控释制剂命名统一命名为:缓释制剂 1号资料(药品名称): B:同A C:同A外,需附上:国家标准 若仿进口药,附上口岸药检报告书即可。(注:不允许申请商品名) 2号资料(证明性文件):
化学试剂的分类和分级 点击次数:567 发布时间:2011-6-1 化学试剂的种类很多,世界各国对化学试剂的分类和分级的标准不尽一致。 IUPAC(International Union of Pure and Applied Chemistry国际理论和应用化学联合会)对化学标准物质的分类为: A级:原子量标准。 B级:和A 级最接近的基准物质。 C级:含量为100+-0.02%的标准试剂 D级:含量为100+-0.05%的标准试剂 E级:以C级或D级为标准对比测定得到的纯度的试剂 化学试剂按用途可分为标准试剂,一般试剂,生化试剂等等。 我国习惯将相当于IUPAC 的C级、D级的试剂称为标准试剂。 优级纯,分析纯,化学纯是一般试剂的中文名称。 一级:即优级纯(GR,Guaranteed reagent);标签为深绿色,用于精密分析试验 二级:即分析纯(AR,Analytical reagent);标签为金光红,用于一般分析试验 三级:即化学纯(CP,Chemical pure)标签为中蓝,用于一般化学试验。 超高纯≥ 99.99% 优级纯≥ 99.8% 分析纯≥ 99.7% 化学纯≥ 99.5% 此外还有其他试剂中文名称,英文简称和英文全名: 化学纯试剂 CP Chemical pure 实验试剂 LR Laboratory reagent 纯 Pur Pure, Purum 特纯 EP Extra Pure
特纯 Puriss Purissimum 精制 Purif Puirfed 生化试剂 BC Biochemical 生物试剂 BR Biological reagent 生物染色剂 BS Biological stain 生物学用 FBP For biological purpose 组织培养用 (无简称) For tissue medium purpose 微生物用 FMB For microbiological 显微镜用 FMP For microscopic purpose 电子显微镜用 (无简称) For electron microscopy 涂镜用 FLB For lens blooming 工业用 Tech Technical grade 实习用 Pract Practical use 分析用 PA Pro analysis 精密分析用 SSG Super special grade 合成用 FS For synthesis 闪烁用 Scint For scintillation 电泳用 (无简称) For electrophoresis use 测折光率用 RI For refractive index 显色剂 (无简称) Developer 研究级 (无简称) Research grade 指示剂 Ind Indicator 络合指示剂 (无简称) Complexon indicator 荧光指示剂 (无简称) Fluorescene indicator 氧化还原指示剂 Redox Redox indicator 吸附指示剂 Adsorb Adsorption indicator 基准试剂 PT Primary reagent 光谱纯 SP Spectrum pure 光谱标准物质 SSS Spectrographic standard substance 分光纯 UV Ultra violet pure 原子吸收光谱 AAS Atomic absorption spectrum 红外吸收光谱 IR Infrared absorption spectrum 核磁共振光谱 NMR Nuclear magnetic resonance spectrum
我国的试剂规格基本上按纯度(杂质含量的多少)划分,共有高纯、光谱纯、基准、分光纯、优级纯、分析和化学纯等7种。国家和主管部门颁布质量指标的主要优级纯、分级纯和化学纯3种。 (1)优级纯(GR: Guaranteed reagent),又称一级品或保证试剂, 99.8%,这种试剂纯度最高,杂质含量最低,适合于重要精密的分析工作和科学研究工作,使用绿色瓶签。 (2)分析纯(AR),又称二级试剂,纯度很高, 99.7%,略次于优级纯,适合于重要分析及一般研究工作,使用红色瓶签。 (3)xx(CP),又称三级试剂,≥ 99.5%,纯度与分析纯相差较大,适用于工矿、学校一般分析工作。使用蓝色(深蓝色)标签。 (4)实验试剂(LR: Laboratory reagent),又称四级试剂。 除了上述四个级别外,目前市场上尚有: 基准试剂(PT: Primary Reagent): 专门作为基准物用,可直接配制标准溶液。 光谱纯试剂(SP: Spectrum pure): 表示光谱纯净。但由于有机物在光谱上显示不出,所以有时主成分达不到 99.9%以上,使用时必须注意,特别是作基准物时,必须进行标定。
纯度xx优级纯的试剂叫做xx试剂(≥ 99.99%)。高纯试剂是在通用试剂基础上发展起来的,它是为了专门的使用目的而用特殊方法生产的纯度最高的试剂。它的杂质含量要比优级试剂低2个、3个、4个或更多个数量级。因此,高纯试剂特别适用于一些痕量分析,而通常的优级纯试剂就达不到这种精密分析的要求。目前,除对少数产品制定国家标准外(如高纯硼酸、高纯冰乙酸、高纯氢氟酸等),大部分高纯试剂的质量标准还很不统一,在名称上有高纯、特纯(Extra Pure)、超纯、光谱纯等不同叫法。根据高纯试剂工业专用范围的不同,可将其分为以下几种: ⑴光学与电子学专用高纯化学品,即电子级试剂(EIectronicgrade)试剂。 ⑵金属-氧化物-半导体(metal-Oxide-Semiconductor)电子工业专用高纯化学品,即UP-S级或MOS试剂(读作: 摩斯试剂)。一般用于半导体,电子管等方面,其杂质最高含量为 0.01-10ppm,有的可降低到ppb数量级,金属杂质含量小于1ppb,尘埃等级达到0-2ppb,适合 0.35— 0.8微米集成电路加工工艺。 ⑶单晶生产用高纯化学品。 ⑷光导纤维用高纯化学品。 此外,还有仪分试剂、特纯试剂(杂质含量低于~级)、特殊高纯度的有机材料等。 (5)等离子体质谱纯级试剂(ICP-Mass Pure Grade): 绝大多数杂质元素含量低于 0.1ppb, 适合等离子体质谱仪(ICP Mass)日常分析工作。
化学药品注册分类新旧版对比 新版(红色字体):NMPA于2020年06月30日发布的《国家药监局关于发布化学药品注册分类及申报资料要求的通告(2020年第44号)》 旧版(蓝色字体):CFDA于2016年03月09日发布的《总局关于发布化学药品注册分类改革工作方案的公告(2016年第51号)》新旧两版均将化药注册分类分为5个类别,具体差异如下: 1类:境内外均未上市的创新药。指含有新的结构明确的、具有药理作用的化合物,且具有临床价值的药品。 1类:境内外均未上市的创新药。指含有新的结构明确的、具有药理作用的化合物,且具有临床价值的原料药及其制剂。 2类:境内外均未上市的改良型新药。指在已知活性成份的基础上,对其结构、剂型、处方工艺、给药途径、适应症等进行优化,且具有明显临床优势的药品。 2类:境内外均未上市的改良型新药。指在已知活性成份的基础上,对其结构、剂型、处方工艺、给药途径、适应症等进行优化,且具有明显临床优势的药品。 2.1含有用拆分或者合成等方法制得的已知活性成份的光学异构体,或者对已知活性成份成酯,或者对已知活性成份成盐(包括含有氢键或配位键的盐),或者改变已知盐类活性成份的酸根、碱基或金
属元素,或者形成其他非共价键衍生物(如络合物、螯合物或包合物),且具有明显临床优势的药品。 2.1含有用拆分或者合成等方法制得的已知活性成份的光学异构体,或者对已知活性成份成酯,或者对已知活性成份成盐(包括含有氢键或配位键的盐),或者改变已知盐类活性成份的酸根、碱基或金属元素,或者形成其他非共价键衍生物(如络合物、螯合物或包合物),且具有明显临床优势的原料药及其制剂。 2.2含有已知活性成份的新剂型(包括新的给药系统)、新处方工艺、新给药途径,且具有明显临床优势的药品。 2.2含有已知活性成份的新剂型(包括新的给药系统)、新处方工艺、新给药途径,且具有明显临床优势的制剂。 2.3含有已知活性成份的新复方制剂,且具有明显临床优势。 2.3含有已知活性成份的新复方制剂,且具有明显临床优势。 2.4含有已知活性成份的新适应症的药品。 2.4含有已知活性成份的新适应症的制剂。 3类:境内申请人仿制境外上市但境内未上市原研药品的药品。该类药品应与参比制剂的质量和疗效一致。 3类:境内申请人仿制境外上市但境内未上市原研药品的药品。该类药品应与原研药品的质量和疗效一致。 具有与参比制剂相同的活性成份、剂型、规格、适应症、给药途径和用法用量,并证明质量和疗效与参比制剂一致。
一、药品研发及注册申报的法规依据 二、药品申请的申报与审批 三、药品的注册分类 四、申报资料的内容 五、药品的监测期 六、药品补充申请注册事项 七、其他配套法规 八、药品研发时企业各部门的配合工作
一、药品研发及注册申报的法规依据 法规依据:《药品注册管理办法》(局令第28号),2007年10月1日起施行。 中国境内申请药物临床试验、药品生产和药品进口,以及进行药品审批、注册检验和监督管理,适用本办法。 药品注册:是指国家食品药品监督管理局(SFDA)根据药品注册申请人的申请,依照法定程序,对拟上市销售药品的安全性、有效性、质量可控性等进行审查,并决定是否同意其申请的审批过程。 国产药品注册申报主要环节:药品研究开发→申报资料的受理(省级药品监督管理局)→研制现场检查并抽取样品(省级/国家药品监督管理局)→抽样的复核检验(省级药检所/中检院)→审评(国家药品审评中心)→审批(国家食品药品监督管理局)。 药品注册申请人:法人机构 药品注册申请分类: 1)新药申请;2)仿制药申请;3)进口药品申请 4)补充申请: 药品申请经批准后,改变、增加或者取消原批准事项或者内容 5)再注册申请:药品批准证明文件有效期满后 二、药品申请的申报与审批 1、研究内容及申报流程(新药两报两批) 1.1临床前研究: 包括药物的合成工艺、提取方法、理化性质及纯度、剂型选择、处方筛选、制备工艺、检验方法、质量指标、稳定性、药理、毒理、动物药代动力学研究等。 1.2 申报临床:获得临床研究批件 1.3临床试验:I、II、III、IV期。 I期:人体耐受程度和药代动力学,20至30例 II期:初步评价治疗作用,100例 III期:确证治疗作用,300例 IV期:上市后应用研究,为2000例 1.4申报生产:获得新药证书及生产批文
附件1-1 化学药品注册分类及申报资料要求 (征求意见稿) 一、化学药品注册分类 化学药品注册分类分为创新药、改良型新药、仿制药、境外已上市境内未上市化学药品,具体分为如下5个类别:1类:境内外均未上市的创新药。指含有新的结构明确的、具有药理作用的化合物,且具有临床价值的药品。 2类:境内外均未上市的改良型新药。指在已知活性成份的基础上,对其结构、剂型、处方工艺、给药途径、适应症等进行优化,且具有明显临床优势的药品。 2.1含有用拆分或者合成等方法制得的已知活性成份的光学异构体,或者对已知活性成份成酯,或者对已知活性成份成盐(包括含有氢键或配位键的盐),或者改变已知盐类活性成份的酸根、碱基或金属元素,或者形成其他非共价键衍生物(如络合物、螯合物或包合物),且具有明显临床优势的药品。 2.2含有已知活性成份的新剂型(包括新的给药系统)、新处方工艺、新给药途径,且具有明显临床优势的药品。 2.3含有已知活性成份的新复方制剂,且具有明显临床优势。 2.4含有已知活性成份的新适应症的药品。 3类:境内申请人仿制境外上市但境内未上市原研药品的药
品。该类药品应与参比制剂的质量和疗效一致。 4类:境内申请人仿制已在境内上市原研药品的药品。该类药品应与参比制剂的质量和疗效一致。 5类:境外上市的药品申请在境内上市。 5.1境外上市的原研药品和改良型药品申请在境内上市。改良型药品应具有明显临床优势。 5.2境外上市的仿制药申请在境内上市。 原研药品是指境内外首个获准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品。 参比制剂是指经国家药品监督管理部门评估确认的仿制药研制使用的对照药品。 二、相关注册管理要求 (一)化学药品1类为创新药,应含有新的结构明确的、具有药理作用的化合物,且具有临床价值,不包括改良型新药中2.1类的药品。含有新的结构明确的、具有药理作用的化合物的新复方制剂,应按照化学药品1类申报。 (二)化学药品2类为改良型新药,在已知活性成份基础上进行优化,应比改良前具有明显临床优势。已知活性成份指境内或境外已上市药品的活性成份。该类药品同时符合多个情形要求的,须在申报时一并予以说明。 (三)化学药品3类为境内生产的仿制境外已上市境内未上市原研药品的药品,具有与参比制剂相同的活性成份、剂型、规格、适应症、给药途径和用法用量,并证明质量和疗效与参比制