
锆—吡咯基配合物的合成与结构:对吡咯基配
体配位方式影响因素的计算分析
□ 文/Joseph M.Tanski ,Gerard Parkin*(约瑟.坦斯基,杰拉德.帕金)
纽约哥伦比亚大学化学学院,纽约10027
2001.11.20 收稿
摘要:具有η1和η5吡咯基配
位方式的锆配合物的一系列结构特点已有初步研究报道。确切的说,2,5 - 二芳基-吡咯[pyr Ar2]配体(Ar= 苯基 ,2,4 – 二甲苯基)已经被用来制备[pyr Ar2]Zr(NMe 2)3(NMe 2H), [pyr Ar2]Zr(NMe 2)3,, [pyr Ar2]Zr(NMe 2)I 2和
[η 5-pyr Ar2]2ZrCl 2.。密度泛函计算结果表明,各种配位方式相对的稳定性可以被立体因素及金属中心的路易斯酸性所影响。
毫无疑问,环戊二烯配
体,在有机过渡金属化学的发展中起到了关键作用。通过和环戊邻二苯基比较,相关等电
子杂环吡咯配体,[pyr Rn ],1已不大适用于过渡金属化学。2关于锆化学,通过X 射线衍射实验,一些吡咯配合物的结构已经得到认证,观察到吡咯配体和戊二烯配体相似,只能通过氮原子结合η1-模式,3而不是η5-模式。在本文中,我们报道了一系列单一和二度(吡咯)锆配合物的合成和结构特征在吡咯配体的η5-配位中起重要的作用,而且计算分析研究致力于找到影响吡咯配体于这些衍生物中η5和η1对抗配位方式的因素。
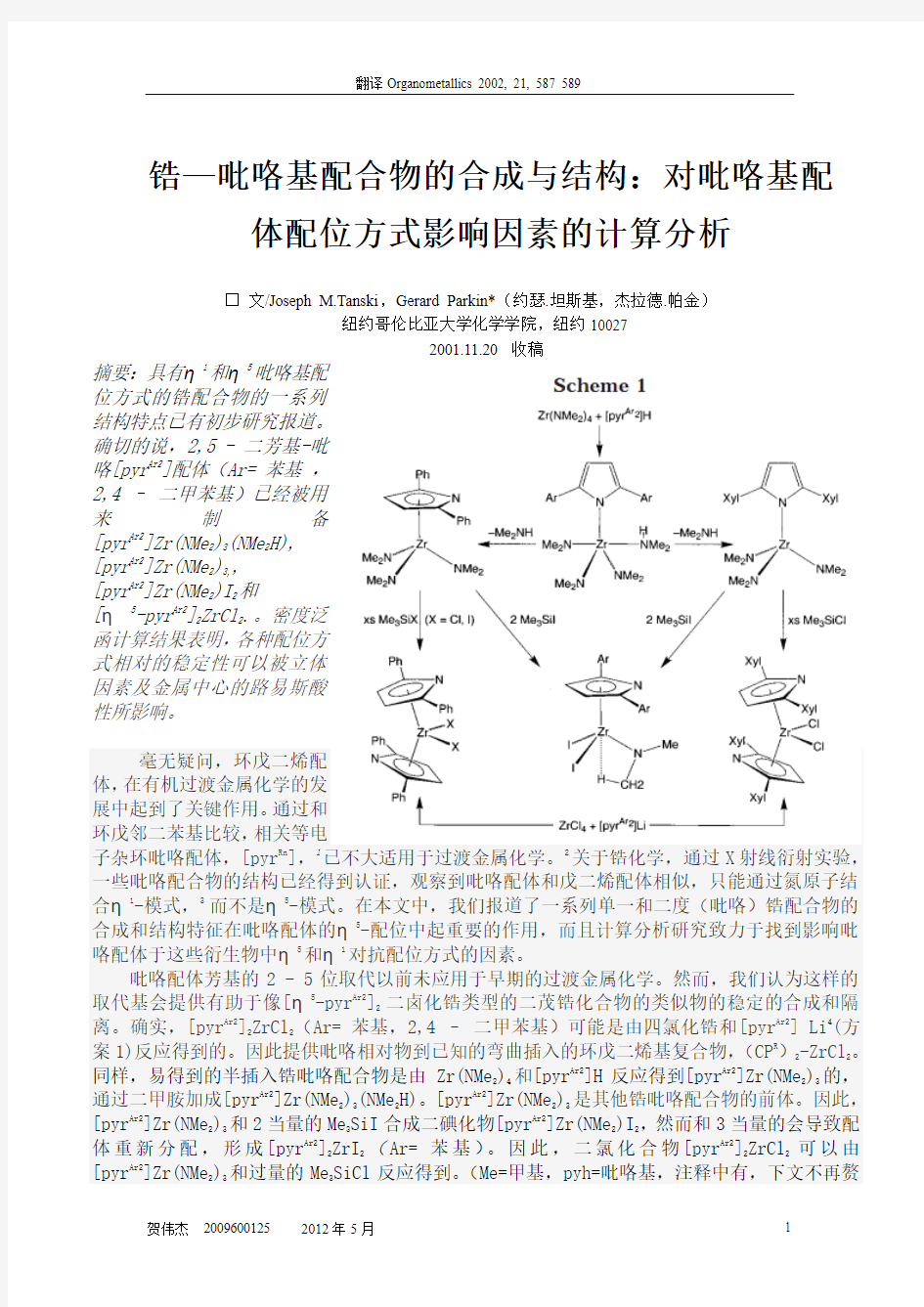
吡咯配体芳基的2 - 5位取代以前未应用于早期的过渡金属化学。然而,我们认为这样的取代基会提供有助于像[η5-pyr Ar2]2二卤化锆类型的二茂锆化合物的类似物的稳定的合成和隔离。确实,[pyr Ar2]2ZrCl 2(Ar= 苯基,2,4 – 二甲苯基)可能是由四氯化锆和[pyr Ar2] Li 4(方案1)反应得到的。因此提供吡咯相对物到已知的弯曲插入的环戊二烯基复合物,(CP R )2-ZrCl 2。同样,易得到的半插入锆吡咯配合物是由 Zr(NMe 2)4和[pyr Ar2]H 反应得到[pyr Ar2]Zr(NMe 2)3的,通过二甲胺加成[pyr Ar2]Zr(NMe 2)3(NMe 2H)。[pyr Ar2]Zr(NMe 2)3是其他锆吡咯配合物的前体。因此,[pyr Ar2]Zr(NMe 2)3和2当量的Me 3SiI 合成二碘化物[pyr Ar2]Zr(NMe 2)I 2,然而和3当量的会导致配体重新分配,形成[pyr Ar2]2ZrI 2(Ar= 苯基)。因此,二氯化合物[pyr Ar2]2ZrCl 2可以由[pyr Ar2]Zr(NMe 2)3和过量的Me 3SiCl 反应得到。(Me=甲基,pyh=吡咯基,注释中有,下文不再赘
述)。
吡咯基配合物配位方式的最终确定需要通过X 射线衍射实验来鉴定(图1-3)。6值得注意的是一个η5吡咯配位方式在苯基取代的吡咯配合物[pyr Ar2]Zr(NMe 2)3,[pyr Ar2]Zr(NMe 2)I 2,[pyr Ar2]2ZrX 2 (X=Cl,I 、Ar= 苯基、pyr=吡咯基) 和对二甲苯基取代取代的吡咯配合物[pyr Xyl 2] 合锆(氮-二甲基)二碘(Xyl=对二甲苯基)和[pyr Xyl 2]2-ZrCl 2.中被找到。鉴于尽管试图具体合成符合η5配位特征7的配合物,但只有对锆的η1配位是预先通过X 射线衍射实验验证的,故η5配位在本系统的观察是最值得注意的。举例来说,配合物Cp 2Zr[η1
-pyr Me2]2和[η1-pyr Me2]4Zr 用加强满足二甲基吡咯配体的立体构型需要将有利于η5配位的概念合成了出来。(图4)但是,η1配位占优和η5配位方式的机理仍然没有掌握3b,8。
尽管在本文中偏向报道锆配合物中的η5配位,但[pyr Ar2]Zr(NMe 2)3(NMe 2H) (Ar =Ph, Xyl)和[pyr Xyl2]Zr(NMe 2)3显示含有η1配位。由于η5配位会因为增加配位数时加大了空间位阻效应从而不占优,那么观察到得二甲胺加成物[pyr Ar2]Zr(NMe 2)3(NMe 2H)的η1配位也变得可以理解。但不是那么容易理解的是二甲苯基衍生物[pyr Xyl2]Zr(NMe 2)3显示含有吡咯配体的
η1配位而苯基衍生物显示含有η5配位确是事实。(图3)这种配位方式上的不同和位阻大的取代基(例如Bu t )在吡咯基的2,5位通常是η5配位优于η1配位2,9,10的概念是相悖的。这样的主张是基于η1配位会迫使其直接向金属中心“弯曲”取代,从而影响相互作用力。位阻更大的[pyr Xyl2]取代基η1配位占优比[pyr Ph2]取代基更显著的现象表明芳基和烷基有不同的取代机理,因为芳基各原子“共平面”。具体来说,邻位取代动摇了吡咯和芳环的共平面构像,这种变化有
助于从η5配位占优到η1
配位占优的转变。如图4所示。
为了进一步解决这个问题,我们通过一系列的密度泛函理论计算来评估η1和η5吡咯基配位方式对[pyr Ar2]Zr(NMe 2)3 和[pyr Ar2]Zr(NMe 2)I 2(Ar=Ph, Xyl)11稳定性的影响。数据表明,η5配位更大程度上倾向于能量更小的配位方式。(i) (NMe 2) I 2取代基比[pyr Xyl2]取代基约少3千卡每摩尔(ii)二碘配合物[pyr Ar2]Zr(NMe 2)I 2比三胺配合物[pyrAr 2]Zr(NMe 2)3约少10千卡每摩尔。增加邻位取代的位阻使[py rXyl2]取代基的η5配位不占优可能是前种配位倾向的合理解释。同样,同时增强NMe 2对碘的位阻效应和给电子能力使在[pyr Ar2]Zr(NMe 2)3中的η5配位比pyr Ar2]Zr(NMe 2)I 2.中更大程度上不占优可能是后种配位倾向的合理解释。
考虑到在[pyr Ph2]Zr(NMe 2)3中η5配位方式比η1配位方式更占优,其钛取代的类似物[pyr Ph2]Ti(NMe 2)3存在吡咯取代基η1配位是值得注意的。此外,计算表明,钛配合物η1配位方式比锆配合物η1配位方式能量高9.25千卡每摩尔,可能是由于钛的位阻小造成的立体相互作用加剧。
[pyr Ar2]Zr(NMe 2)3和[py rAr2]Zr(NMe 2)I 2衍生物之间一个有趣的区别是,二碘配合物NMe 2配体的甲基和金属中心锆之间存在β-抓氢键。NMe 2配体在平面三角几何上的显著变形为它的这种罕
见的
键提供了证据。13例如,[pyr Ph2]Zr(NMe
2)I
2
中Zr-N-Me的键角分别为108.9(8)°和141.3(6)°,
这和剑桥结构数据库中列出的锆-二甲氨基配合物的结构特征平均键角为125.7°的数值有很大的差别。14短键 Zr?···?C之间(2.84(1) ?)和Zr?····H之间 (2.61(8)?)的键距也和抓氢键的
键距相符。例如,[pyr ph2]Zr(NMe
2)I
2
中Zr????···?C的键距介于它在阳离子氢键乙基配合物
{[Cp Me]
2Zr(Et)(PMe
3
)}+的键距(2.63 ?)15和它在九氢键乙基配合物中的键距(3.17 3.42 ?).14之
间。
鉴于锆-配合物在烯烃聚合方面的广泛应用,[pyr Ph2]ZrCl
2
在methylalumoxane甲基矾环氧
乙烷(MAO)中的催化活性已有研究。实际上Cp
2ZrCl
2
/MAO的烯烃聚合催化效率约是
[pyr Ph2]ZrCl
2
/MAO的20倍。
总的来说,一系列含有[pyr Ar2]配体的η1和η5吡咯配位方式的锆-配合物已经制得。密度泛函理论(DET)计算表明,各种配位方式的相对稳定性受立体因素和金属中心的路易斯酸性共同影响,如此以致大位阻和π电子供体配体在接近金属中心时倾向η1配位方式。除了位阻效应以外,芳基的邻位取代也会降低η5配位方式的相对稳定性,因为它破坏了吡咯和芳环的共平面构象。这样,增大芳基的位阻效应可能会导致与烷基η1配位方式比η5配位方式占优相反的结论。本文报道的结构表明,被取代的吡咯配体提供了影响锆-配合物配位立体结构多样的途径。
注释:
1.[pyr Rn]是吡咯配体(C
5RnH
4-n
N)的缩写,其中上标表示取代基的数量和类型。
2.参考文献:(a) Kershner, D. L.; Basolo, F. Coord.《化学发现》1987
年第79期279到292页;(b) Nief, F. Eur. J.《无机化学》2001年891页到904页。
3.参考文献:(a) Bynum, R. V.; Hunter, W. E.; Rogers, R. D.; Atwood,
J. L. 《无机化学》1980年第19期2368到2374页;(b) Bynum, R. V.;
Zhang, H. M.;Hunter, W. E.; Atwood, J. L. 《Can. J. Chem.》1986年64期1304到1307页。
4.[pyr Xyl2]H使用合成[pyr Ph2]H相似的方法合成的,见于Patterson, J. M.;
Soedigdo, S. J.《有机化学》1968年第33期2057到2061页。
5.[pyr Ph2]Zr(NMe
2)I
2
也能由[pyr Ph2]Zr(NMe
2
)
3
和4当量的MeI在Me
4
NI是副产
物的反应中合成,此外,碘化物[pyr Ph2]Zr(NMe
2)I
2
中的碘来自2当量的
MeI。
6.见晶体结构数据的支持信息。
7.然而值得注意的是,吡咯片段对锆中心的η5配位方式已经在卟啉衍生物
中有了观察研究。见于,比如(a) Solari, G.; Solari, E.; Floriani,
C.;Chiesi-Villa, A.; Rizzoli, C.《有机金属》1997年第16期508到
510页;Jacoby, D.; Floriani, C.; Chiesi-Villa, A.; Rizzoli, C.
《JACS》1993年115期3595到3602页。
8.对于锆-吡咯配合物,它们的配位方式还没有经X射线衍射实验所证实。
见于(a) Kuhn, N.;Stubenrauch, S.; Boese, R.; Bl?ser, D.
《https://www.doczj.com/doc/642528567.html,anomet. Chem》1992年440期289到296页;(b) Dias, A. R.;
Galv?o, A. M.; Galv?o,A.C. 《https://www.doczj.com/doc/642528567.html,anomet. Chem》2001年632期157到163页。
9.参考文献:Bynum, R. V.; Zhang, H. M.; Hunter, W. E.; Atwood, J. L.
《Can. J. Chem.》1986年64期1304到1307页。
10.参考文献:(a) Dias, A. R.; Galv?o, A. M.; Galv?o, A. C.; Salema,
M. S.《J.Chem. Soc.》道尔顿翻译1997年1055到1061页;(b) Dias,
A. R.; Galv?o,A. M.; Galv?o,A.C.《Collect. Czech. Chem. Commun.》
1998年63期182到186页。
11.3-ζ基组cc-pVTZ (-f)在B3LYP水平上能计算除了Ti, Zr, Cl, 和 I之
外的所有元素的单点能量优化结构,所以密度泛函理论计算的几何优化在LACVP**基组的B3LYP水平上进行。(Jaguar 4.1, Schrodinger, Inc.,Portland, OR, 2000).
12.Δ E =E(η5)-E(η1)的值(单位kcal mol-1)[pyr Ph2]Zr(NMe
2) 3
(1.76),[pyr Xyl2]Zr(NMe
2)
3
(4.61), [pyr Ph2]Zr(NMe
2
)I
2
(-9.15),
[pyr Xyl2]Zr(NMe
2)I
2
(-5.73).
13.虽然少见,但是查阅剑桥结构数据库发现其他几个二甲氨基-锆配合物表
现出这种键。见于,例如: (a) Chisholm, M.H.; Hammond, C. E.; Huffman, J. C.《 Polyhedron》 1988年, 7期, 2515-2520页.(b) Oberthu ¨ r, M.; Hillebrand, G.; Arndt, P.; Kempe, R. 《Chem. Ber./Recl.》 1997年, 130期, 789-794页。
14.参考文献:Allen, F. H.; Kennard, O. CSD 版本5.21. 3D用剑桥结构
数据库检索,《Chem. Design Automation News 》1993年, 8(1), 1, 31-37页.
15.参考文献:Jordan, R. F.; Bradley, P. K.; Baenziger, N. C.; LaPointe,
R. E.J. Am.《 Chem. Soc. 》1990年, 112期, 1289-1291页。
学习体会:此篇论文与配位化学课本中的价键理论,配合物的合成与结构,X衍射晶体结构分析,配合物的反应及催化性能等都有联系,结合课本我对本文也有了一定的了解,在了解的基础上进行了翻译。当然,由于水平的问题,很多地方翻译得不是很妥当,有些地方还有错误。此篇论文让我印象最深的是关于对配合物异构现象的深入讨论,对影响其构象的因素进行了计算分析,然后得出了结论。我学到的不仅是知道了影响各种配位方式相对的稳定性的是立体因素及金属中心的路易斯酸性,更是这种研究方法本身。先是发现一个现象,对这个现象进行讨论,并用计算的方法得出结论,这是严谨科学的方法。
本文的创新点:本文用计算分析的方法对影响对影响配位方式的因素进行了讨论。创新的地方是对特殊的键的讨论,得出了可能的原因。数学建模后的计算分析。
实验五 配合物的生成和性质 一、实验目的 1、了解有关配合物的生成,配离子及简单离子的区别。 2、比较配离子的稳定性,了解配位平衡与沉淀反应、氧化还原反应以及溶液酸度的关系。 二、预习提问 1、 试指出硫酸铁铵和铁氰化钾哪个物质是配合物? 答:铁氰化钾是配合物。 2、 Cu 2+与[Cu (NH 3)4]2+比,谁的氧化能力较强?为什么? 答:Cu 2+的氧化能力较强。因为φCu2+/Cu >φ[Cu (NH3)4]2+/Cu 3、 配合物与复盐的主要区别是什么?如何判断某化合物是配合物? 答:复盐是由两种或两种以上的同种晶型的简单盐类所组成的化合物,在其晶体中(或水溶液中)均只有简单的离子存在。而配合物晶体中存在复杂的配位离子或配位分子,这些配位离子或配位分子或离子很稳定,能以独立的整体存在,根据晶体中(或水溶液中)是否含有复杂的配位离子或分子来判断某化合物是不是配合物。 三、实验原理 由一个简单的正离子和几个中性分子或其它离子结合而成的复杂离子叫配离子,含有配离子的化合物叫配合物。配离子在溶液中也能或多或少地离解成简单离子或分子。例如: [Cu(NH 3)4]2+配离子在溶液中存在下列离解平衡: 32243NH 4Cu ])NH (Cu [+?++ )])(([)()(243342++?=NH Cu C NH C Cu C K d 不稳定常数K d 表示该离子离解成简单离子趋势的大小。 配离子的离解平衡也是一种化学平衡。能向着生成更难离解或更难溶解的物质的方向进行,例如,在[Fe(SCN)]2+溶解中加入F -离子,则反应向着生成稳定常数更大的[FeF 6]3- 配离子方向进行。 螯合物是中心离子与多基配位形成的具有环状结构的配合物。很多金属的螯合物都具有特征的颜色,并且很难溶于水而易溶于有机溶剂。例如,丁二肟在弱碱性条件下与Ni 2+生成鲜红色难溶于水的螯合物,这一反应可作检验Ni 2+的特征反应。 四、仪器及试剂 1、 仪器 试管、滴定管 2、 试剂 HgCl 2(0.1mol ·L -1)、KI(0.1 mol ·L -1)、NiSO 4(0.2 mol ·L -1)、BaCl 2(0.1mol ·L -1)、NaOH(0.1mol ·L -1)、1:1(NH 3·H 2O)、FeCl 3(0.1mol ·L -1)、KSCN(0.1 mol ·L -1)、K 3[Fe(CN)6](0.1 mol ·L -1)、AgNO 3(0.1mol ·L -1)、NaCl(0.1 mol ·L -1)、CCl 4、FeCl 3(0.5 mol ·L -1)、NH 4F(4 mol ·L -1)、
第十八章配位化合物的价键理论 §本章摘要§ 1.配位化合物的基本概念 配位化合物配合物的命名异构现象 2.配位化合物的稳定性 酸碱的软硬分类影响配位单元稳定的因素 3.配合平衡 配合-解离配合配合平衡的移动 4.配位化合物的价键理论 配合物的构型与中心的杂化方式中心杂化轨道的形成价键理论中的能量问题价键理论的实验根据 5.配合物的晶体场理论 晶体场中的 d 轨道过渡金属化合物的颜色晶体场稳定化能(CFSE)Jahn - Teller 效应 §1.配位化合物的基本概念 一.配位化合物 1 定义 由中心原子( 或离子) 和几个配体分子(或离子)以配位键相结合而形成的复杂分子或离子,通常称为配位单元。凡是含有配位单元的化合物都称做配位化合物,简称配合物,也叫络合物。 , Ni(CO)4都是配位单元,分别称作配阳离子、配阴离子、配分子。 [ Co(NH3)6]Cl3 , K3[Cr(CN)6], Ni(CO)4都是配位化合物。[Co(NH3)6] [Cr(CN)6] 也是配位化合物。判断的关键在于是否含有配位单元。 2 构成 内界是配位单元,外界是简单离子。 又如K3[Cr(CN)6] 之中,内界,外界是K+。可以无外界,如Ni(CO)4。但不能没有内界。内外界之间是完全电离的。 内界配位单元又由中心和配体构成。中心:又称为配合物的形成体,多为金属离子,尤其是过渡金属离子。配体:经常是阴离子或分子。 3 配位原子和配位数 配体中给出孤对电子与中心直接形成配位键的原子,叫配位原子。配位单元中,中心周围与中心直接成键的配位原子的个数,叫配位数。 配位化合物[Co(NH3)6]Cl3的内界为,中心的周围有6 个配体NH3,每个NH3中有1 个N 原子与配位。N 是配位原子,Co 的配位数是6 。注意:配体的个数与配位数不是同一个概念。 若中心的电荷高,半径大,则利于形成高配位数的配位单元;而配体的电荷高,半径大,利于低配位数。 4 多基配体和螯合物
配合物的化学键理论 摘要:化学键理论在配位化学中有着重要的运用,它现在主要有三大流派。本文就回顾化学键的发展历程,并对三大化学键理论做出仔细的阐述。 关键字:化学键价键理论分子轨道理论晶体场理论配位场理论 十八世纪后半叶,欧洲的化学家开始了定量的化学实验的研究。法国化学家普劳斯特通过测定部分化合物的重量组成而提出了定组成定律即一个化合物不管它是天然的还是人工合成的组成该化合物的各元素的重量百分比是固定不变的这一定律促使人们进一步研究化合物是怎样组成的和靠什么力结合在一起的。化合物的定组成结构和性质有什么关系。由此化学键理论产生和逐步发展起来。 1 化学键的发展历程 最早化学家假设原子和原子之间是用一个神秘的钩钩住的,这种设想至今仍留下痕迹,化学键的“键”字就有钩的意思。 1916年,德国科学家柯塞尔考察大量的事实后得出结论:任何元素的原子都要使最外层满足8 电子稳定结构。柯塞尔的理论能解释许多离子化合物的形成,但无法解释非离子型化合物。1923 年,美国化学家路易斯发展了柯塞尔的理论,提出共价键的电子理论:两种元素的原子可以相互共用一对或多对电子,以便达到稀有气体原子的电子结构,这样形成的化学健叫做共价健。 柯塞尔和路易斯的理论常叫原子价电子理论。它只能定性地描述分子的形成,化学家更需要对化学键做定量阐述。 1927 年,海特勒和伦敦用量子力学处理氢分子,用近似方法计算出氢分子体系的波函数和能量获得成功,这是用量子力学解决共价键问题的首例。1930 年,鲍林更提出原子成键的杂化理论(杂化轨道理论)。1932 年,洪德把单键、多键分成δ和∏键两类。δ健是指在沿着连接两个原子核的直线(对称轴)上电子云有最大重叠的共价键,这种键比较稳定。∏键是指沿电子云垂直于这条直线方向上结合而成的键,这种键比较活泼。这就使价键理论进一步系统化,使经典的化合价和化学键有机地结合在一起了。 由于上述的价键理论对共扼分子、氧气分子的顺磁性等事实不能有效解释,因此本世纪30 年代后又产生一种新的理论——分子轨道理论。 分子轨道理论在1932 年首先由美国化学家马利肯提出。他用的方法跟经典化学相距很远,一时不被化学界接受,后经密立根、洪德、休克尔、伦纳德等人努力,使分子轨道理论得到充实和完善。它把分子看作一个整体,原子化合成分子时,由原子轨道组合成分子轨道,
化学前沿 【论文摘要】: 无机化学是化学学科里其它各分支学科的基础学科,在近年来取得较突出的进展,主要表现在固体材料化学、配位化学等方面。未来无机化学的发展特点是各学科交叉纵横相互渗透,用以解决工业生产与人民生活的实际问题。文章就当代无机化学研究的前沿与未来发展趋势做了简要阐述。 当前无机化学的发展趋向主要是新型的无机化合物的合成和应用,以及新的研究领域的开辟和建立。因此21世纪理论与计算方法的运用将大大加强理论和实验更加紧密的结合。同时各学科间的深入发展和学科间的相互渗透,形成许多学科的新的研究领域。例如,生物无机化学就是无机化学与生物学结合的边缘学科;固体无机化学是十分活跃的新兴学科;作为边沿学科的配位化学日益与其它相关学科相互渗透与交叉。 根据国际上最新进展和我国的具体情况,文章就“无机合成与制备化学研究进展”和“我国无机化学最新研究进展”两个方面进行阐述: 一、无机合成与制备化学研究进展 无机合成与制备在固体化学和材料化学研究中占有重要的地位, 是化学和材料科学的 基础学科。发展现代无机合成与制备化学, 不断地推出新的合成反应和路线或改进和绿化现有的陈旧合成方法, 不断地创造与开发新的物种, 将为研究材料结构、性能(或功能) 与反应间的关系、揭示新规律与原理提供基础。近年来无机合成与制备化学研究的新进展主要表现为以下几个方面: (一)极端条件合成 在现代合成中愈来愈广泛地应用极端条件下的合成方法与技术来实现通常条件下无法进行的合成, 并在这些极端条件下开拓多种多样的一般条件下无法得到的新化合物、新物相与物态。超临界流体反应之一的超临界水热合成就是无机合成化学的一个重要分支。 (二)软化学合成 与极端条件下的合成化学相对应的是在温和条件下功能无机材料的合成与晶化, 即温 和条件下的合成或软化学合成。由于苛刻条件对实验设备的依赖与技术上的不易控制性, 减弱了材料合成的定向程度。而温和条件下的合成化学——即“软化学合成”,正是具有对实验设备要求简单和化学上的易控性和可操作性特点, 因而在无机材料合成化学的研究领域中 占有一席之地。 (三)缺陷与价态控制 缺陷与特定价态的控制是固体化学和固体物理重要的研究对象, 也是决定和优化材料 性能的主要因素。材料的许多性质如发光、导电、催化等都和缺陷与价态有关。晶体生长行为和材料的反应性与缺陷关系密切, 因此, 缺陷与价态在合成中的控制显然成为重要的科学题。缺陷与特定价态的生成和变化与材料最初生成条件有关, 因此,可通过控制材料生成条件来控制材料中的缺陷和元素的价态。 (四)计算机辅助合成 计算机辅助合成是在对反应机理有了了解的基础上进行的理论模拟过程。国际上一般为建立与完善合成反应与结构的原始数据库, 再在系统研究其合成反应与机理的基础上, 应用神经网络系统并结合基因算法、退火、mon te2carlo 优化计算等建立有关的合成反应数学模型与能量分布模型, 并进一步建立定向合成的专家决策系统。
配位化学基础 配位化学就是在无机化学基础上发展起来得一门具有很强交叉性得学科,配位化学旧称络合物化学,其研究对象就是配合物得合成、结构、性质与应用。配位化学得研究范围,除最初得简单无机加与物外,已包括含有金属-碳键得有机金属配位化合物,含有金属-金属键得多核蔟状配位化合物即金属簇合物,还包括有机配体与金属形成得大环配位化合物,以及生物体内得金属酶等生物大分子配位化合物。 一、配合物得基本概念 1、配合物得定义及构成 依据1980年中国化学会无机化学命名原则,配合物可以定义为:由可以给出孤对电子或多个不定域电子得一定数目得离子或分子(统称为配体)与具有接受孤对电子或多个不定域电子得空位得原子或离子(统称为中心原子),按一定得组成与空间构型所形成得化合物。结合以上规定,可以将定义简化为:由中心原子或离子与几个配体分子或离子以配位键相结合而形成得复杂分子或离子,统称为配体单元。含配体单元(又称配位个体)得化合物称为配位化合物。 配体单元可以就是配阳离子,配阴离子与中性配分子,配位阳离子与阴离子统称配离子。配离子与与之平衡电荷得抗衡阳离子或阴离子结合形成配位化合物,而中性得配位单元即时配位化合物。但水分子做配体得水合离子也经常不瞧成配离子。 配位化合物一般分为内界与外界两部分,配体单元为内界,抗衡阳离子或阴离子为外界,而含中性配位单元得配位化合物则无外界。配合物得内界由中心与配体构成,中心又称为配位化合物得形成体,多为金属,也可以就是原子或离子,配体可以就是分子、阴离子、阳离子。 2、配位原子与配位数 配位原子:配体中给出孤对电子与中心直接形成配位键得原子 配位数:配位单元中与中心直接成键得配位原子得个数配位数一般为偶数,以4、6居多,奇数较少 配位数得多少与中心得电荷、半径及配体得电荷、半径有关: 一般来说,中心得电荷高、半径大有利于形成高配位数得配位单元,如氧化数为+1得中心易形成2配位,氧化数为+2得中心易形成4配位或6配位,氧化数为+3得易形成6配位。配体得半径大,负电荷高,易形成低配位得配位单元。 配位数得大小与温度、配体浓度等因素有关: 温度升高,由于热震动得原因,使配位数减少;配体浓度增大,利于形成高配位。
配合物的生成和性质 一、实验目的 1、比较并解释配离子的稳定性; 2、了解配位离解平衡与其它平衡之间的关系; 3. 了解配合物的一些应用。 二.实验原理 中心原子或离子与一定数目的中性分子或阴离子以配位键结合形成配位个体。配位个体处于配合物的内界。若带有电荷就称为配离子,带正电荷称为配阳离子,带负电荷称为配阴离子。配离子与带有相同数目的相反电荷的离子(外界)组成配位化合物,简称配合物。 简单金属离子在形成配离子后,其颜色,酸碱性,溶解性及氧化还原性都会变化。配离子之间也可转化,又一种配离子转化为另一种稳定的配离子。 具有环状结构的配合物称为螯合物,螯合物的稳定性更大,且具有特征颜色。三.实验内容 1. 简单离子与配离子的区别 铁氰化钾K3[Fe(CN)6]加SCN-无血红色 Fe3+ + nSCN- = [Fe(NCS)n]3-n有血红色 结论:FeCl3为离子型简单化合物,在水中可解离出大量的Fe3+,K3[Fe(CN)6]为配合物,配离子[Fe(CN)6]3-比较稳定,难以解离出大量的Fe3+。 2. 配离子稳定性的比较 (1) Fe3+ + n SCN- = [Fe(NCS)n]3-n(有血红色) [Fe(NCS)n]3-n+(C2O4)22-→[Fe(C2O4)3]3-+SCN-
稳定性[Fe(C2O4)3]3->[Fe(NCS)n]3-n (2)AgNO3+ NaCl →AgCl ↓(白) + NH3?H2O→ [Ag(NH3)2]+ [Ag(NH3)2]++ KBr →AgBr↓(淡黄色), 再滴加Na2S2O3溶液→ 沉淀溶解[Ag(S2O3)2]3-,滴加KI溶液→AgI↓AgBr + 2S2O32-→[Ag(S2O3)2]3- + Br-; [Ag(S2O3)2]3- + I-→AgI↓+ 2S2O32-比较:K SP?: AgCl>AgBr>AgI; 稳定性: [Ag(S2O3)2]3-> [Ag(NH3)2]+ (3) I2 + [Fe(CN)6]4- = I- + [Fe(CN)6]3- E? (Fe3+/Fe2+) > E? (I2/I-) > E? ([Fe(CN)6]3-/[Fe(CN)6]4-) 稳定性[Fe(CN)6]3->[Fe(CN)6]4- 3. 配位离解平衡的移动 2CuSO4 + 2NH3·H2O → Cu2(OH)2SO4↓+(NH4)2SO4 Cu2(OH)2SO4↓+8NH3·H2O→ [Cu(NH3)4]SO4 + [Cu(NH3)4](OH)2 + 4H2O (1) 利用酸碱反应破坏[Cu(NH3)4]2+ SO42- + 2[Cu(NH3)4]2+ + 6H+ + 2H2O = Cu2(OH)2SO4↓ + 8NH4+ (2) 利用沉淀反应破坏[Cu(NH3)4]2+ [Cu(NH3)4]2+ + S2-→ CuS↓ + 4NH3 (3) 利用氧化还原反应破坏[Cu(NH3)4]2+ [Cu(NH3)4]2+ + Zn = [Zn(NH3)4]2+ + Cu (4) 利用生成更稳定配合物(螯合物)的方法破坏[Cu(NH3)4]2+ [Cu(NH3)4]2++ edta4-→[Cu(edta)]2-+ 4NH3 4. 配合物的某些应用 (1) 利用生成有色配合物定性鉴定某些离子 pH控制为5-10:Ni2+ + NH3?H20 +二乙酰二肟→ 鲜红色沉淀
1 配位化学导论总结 1. 配位化学 1) 定义:金属或金属离子同其他分子或离子相互结合的化学。 2) 基础:无机化学 3) 重要性:与其他学科互相渗透的交叉性学科 4) 发展: ● 近代配位化学: “键理论”等理论无法全面说明形成机理与成键方式. ● 现代配位化学理论:建立:1893年,瑞士化学家维尔纳提出了现代的配位键、配位数和配位化合物结构的基本概念,并用立体化学观点成功地阐明了配合物的空间构型和异构现象。 2. 配合物的基本概念 1) 定义:由具有接受孤对电子或多个不定域电子的空位原子或离子(中心体)与可以给出孤对电子或多个不定域电子的一定数目的离子或分子(配体)按一定的组成和空间构型所形成的物种称为配位个体,含有配位个体的化合物成为配合物。 2) 组成: 内界、外界、中心体、配体、配位原子 3) 配体分类: 4) 中心原子的配位数: ● 定义:单齿配体:配位数等于内界配体的总数。多齿配体:各配体的配位原子数与配体个数乘积之和。 ● 影响中心原子的配位数因素: A 、按配 体所含配 位原子的 数目分两 种: B 、根据 键合电子 的特征分 为三种:
3. 配合物的分类 4. 配合物的命名 原则是先阴离子后阳离子,先简单后复杂。 一、简单配合物的命名: (1)先无机配体,后有机配体 cis - [PtCl2(Ph3P)2] 顺-二氯 二?(三苯基磷)合铂(II) (2) 先列出阴离子,后列出阳离子,中性分子(的名称) K[PtCl3NH3] 三氯?氨合铂(II)酸钾 (3) 同类配体(无机或有机类)按配位原子元素符号的英文字母顺序排列。 [Co(NH3)5H2O]Cl3 三氯化五氨?一水合钴(III) 中心离子 对配位数 的影响 配体对配 位数的影 响1、按中心原 子数目分为: 2、按配合物 所含配体种 类分为: 3、按配体的 齿数分类: 4、按配合物 地价键特点 分类:
第二章配合物的化学键 理论 内容:研究中心原子和配体之间结合力的本性。 目标:解释性质,如配位数、几何结构、磁学性质、光谱、热力学稳定性、动力学反应性等。 三种理论:①价键理论、②晶体场理论、③分子轨道理论 第一节价键理论(Valence bond theory) 由L.Pauling提出 一、理论要点: ①配体的孤对电子可以进入中心原
子的空轨道;中心原子总是用空轨道杂化,然后用杂化轨道接收配体提供的孤对电子。 ②中心原子用于成键的轨道是杂化轨道(用于说明构型)。中心原子的价层电子结构与配体的种类和数目共同决定杂化类型。 ③杂化类型决定配合物的空间构型,磁距和相对稳定性。 二、轨道杂化及对配合物构型的解释 能量相差不大的原子轨道可通过线性组合构成相同数目的杂化轨道。 对构型的解释(依据电子云最大
重叠原理:杂化轨道极大值应指向配体) 指向实例 sp3、sd3杂化四面体顶点Ni(CO)4 sp2、sd2、dp2、d3杂化三角形顶点[AgCl3]2- dsp2、d2p2 杂化正方形顶点[PtCl4]2- d2sp3杂化八面体顶点[ Fe(CN)6]4- sp杂化直线型[AgCl2]-
三、内轨型和外轨型 若要形成ML6型配合物(L为单齿配体),则需要6个空杂化轨道接收6个L提供的孤电子对,满足该条件的杂化类型有d2sp3和sp3 d2。尽管这两种杂化都导致八面体型配合物,但前者是次外层(n-1)d轨道,而后者是最外层nd轨道,因此与这两种杂化相应的配合物分别称内轨型和外轨型配合物。 中心原子的价层电子数和配体的性质都是影响配合物内轨型和外轨型的因素。当d电子数≤3时,该层空d轨道≥2,总是生成内轨型配合物。
当代无机化学研究前沿与进展 【摘要】: 无机化学是化学学科里其它各分支学科的基础学科,在近年来取得较突出的进展,主要表现在固体材料化学、配位化学等方面。未来无机化学的发展特点是各学科交叉纵横相互渗透,用以解决工业生产与人民生活的实际问题。文章就当代无机化学研究的前沿与未来发展趋势做了简要阐述。 【关键词】:无机化学;研究前沿;研究进展 当前无机化学的发展趋向主要是新型的无机化合物的合成和应用,以及新的研究领域的开辟和建立。因此21世纪理论与计算方法的运用将大大加强理论和实验更加紧密的结合。同时各学科间的深入发展和学科间的相互渗透,形成许多学科的新的研究领域。例如,生物无机化学就是无机化学与生物学结合的边缘学科;固体无机化学是十分活跃的新兴学科;作为边沿学科的配位化学日益与其它相关学科相互渗透与交叉。 根据国际上最新进展和我国的具体情况,文章就“无机合成与制备化学研究进展”和“我国无机化学最新研究进展”两个方面进行阐述: 一、无机合成与制备化学研究进展 无机合成与制备在固体化学和材料化学研究中占有重要的地位, 是化学和材料科学的基础学科。发展现代无机合成与制备化学, 不断地推出新的合成反应和路线或改进和绿化现有的陈旧合成方法, 不断地创造与开发新的物种, 将为研究材料结构、性能(或功能) 与反应间的关系、揭示新规律与原理提供基础。近年来无机合成与制备化学研究的新进展主要表现为以下几个方面: (一)极端条件合成 在现代合成中愈来愈广泛地应用极端条件下的合成方法与技术来实现通常条件下无法进行的合成, 并在这些极端条件下开拓多种多样的一般条件下无法得到的新化合物、新物相与物态。超临界流体反应之一的超临界水热合成就是无机合成化学的一个重要分支。 (二)软化学合成 与极端条件下的合成化学相对应的是在温和条件下功能无机材料的合成与晶化, 即温和条件下的合成或软化学合成。由于苛刻条件对实验设备的依赖与技术上的不易控制性, 减弱了材料合成的定向程度。而温和条件下的合成化学——即“软化学合成”, 正是具有对实验设备要求简单和化学上的易控性和可操作性特点, 因而在无机材料合成化学的研究领域中占有一席之地。
第27章配位化合物(01) 27.1 配位层的特性 27.1 共价键和配位共价键的区别是什么?在NH4+离子中分别有多少 个共价键和配位共价键?如何对其进行区分? 解配位共价键是指一对电子由两个原子共享,且此电子是由其中的一个原子提供的;共价键是指一对共用电子对,一旦形成这两种键就没有区别。在NH4+离子中有四个共价键,其中有一个是配位共价键。 27.2 八面体共有几个面?几个角?具有八面体配位结构的中心离子 的配位数是多少? 解顾名思义八面体有八个面,但有六个角。因为配位体处于八面体的角上,所以具有八面体配位结果的金属的配位数是六。 27.3 在无限稀的溶液中CoBr·4NH3·2H2O的摩尔电导率为: 420cm-1·Ω-1,由此推导此配位化合物的组成。 解此电导率与(3+,1-)的电解质相对应;所以配位化合物可表示为:[Co(NH3)4(H2O)2]3+(Br-)3,或为更简单的:[Co(NH3)4(H2O)2]Br3。 27.4 求下列配位化合物的中心原子的配位数分别是多少? (a) [Mo(CN)8]4-中的钼(b)Cu(en)22+中的铜(en为乙二胺) 解(a) 8 (b) 4 27.5 配平方程式:AgCl(s)+NH3→ 解AgCl(s)+NH3 → Au(NH3)2++Cl- 27.6 把下列各物质按摩尔电导率递增的顺序排列:(a) K[Co(NH3)2(NO2)4] (b) Cr(NH3)3(NO2)3] (c) [Cr(NH3)5(NO2)]3[Co(NO2)6]2(d) Mg[Cr(NH3)(NO2)5] 解离子数及其电荷数越多其电导率越大,则按摩尔电导率递增的顺序排列为:b 配位化学基础 配位化学是在无机化学基础上发展起来的一门具有很强交叉性的学科,配位化学旧称络合物化学,其研究对象是配合物的合成、结构、性质和应用。配位化学的研究范围,除最初的简单无机加和物外,已包括含有金属-碳键的有机金属配位化合物,含有金属-金属键的多核蔟状配位化合物即金属簇合物,还包括有机配体与金属形成的大环配位化合物,以及生物体内的金属酶等生物大分子配位化合物。 一、配合物的基本概念 1.配合物的定义及构成 依据1980年中国化学会无机化学命名原则,配合物可以定义为:由可以给出孤对电子或多个不定域电子的一定数目的离子或分子(统称为配体)和具有接受孤对电子或多个不定域电子的空位的原子或离子(统称为中心原子),按一定的组成和空间构型所形成的化合物。结合以上规定,可以将定义简化为:由中心原子或离子和几个配体分子或离子以配位键相结合而形成的复杂分子或离子,统称为配体单元。含配体单元(又称配位个体)的化合物称为配位化合物。 配体单元可以是配阳离子,配阴离子和中性配分子,配位阳离子和阴离子统称配离子。配离子与与之平衡电荷的抗衡阳离子或阴离子结合形成配位化合物,而中性的配位单元即时配位化合物。但水分子做配体的水合离子也经常不看成配离子。 配位化合物一般分为内界和外界两部分,配体单元为内界,抗衡阳离子或阴离子为外界,而含中性配位单元的配位化合物则无外界。配合物的内界由中心和配体构成,中心又称为配位化合物的形成体,多为金属,也可以是原子或离子,配体可以是分子、阴离子、阳离子。 2.配位原子和配位数 配位原子:配体中给出孤对电子与中心直接形成配位键的原子 配位数:配位单元中与中心直接成键的配位原子的个数配位数一般为偶数,以4、6居多,奇数较少 配位数的多少和中心的电荷、半径及配体的电荷、半径有关: 一般来说,中心的电荷高、半径大有利于形成高配位数的配位单元,如氧化数为+1的中心易形成2配位,氧化数为+2的中心易形成4配位或6配位,氧化数为+3的易形成6配位。配体的半径大,负电荷高,易形成低配位的配位单元。 配位数的大小与温度、配体浓度等因素有关: 温度升高,由于热震动的原因,使配位数减少;配体浓度增大,利于形成高配位。 配位数的大小与中心原子价电子层结构有关: 价电子层空轨道越多一般配位数较高 配位数的大小与配体位阻和刚性有关: 配体的位阻一般都会使中心原子的配位数降低,位阻越大、离中心原子越近,配位数的降低程度也就越大。配体的刚性不利于配体在空间中的取向,长回事中心原子的配位数降低。 3.配体的类型 现代配位化学的研究领域及配位化学的应用现代配位化学既有理论又有事实,它把最新的量子力学成就作为自己阐述配合物性质的理论基础, 也力图用热力学、动力学的知识去揭示配位反应的方向 和历程。 已经进入到了现代发展阶段的现代配位化学具有如下三个特点: ●从宏观到微观 现代配位化学进入到物质内部层次的研究阶段,也即进入了微观水平的研 究阶段。现在不只研究配位化合物的宏观性质,而且更重视物质微观结构的研 究即原子、分子内部结构特别是原子、分子中电子的行为和运动规律的研究, 从而建立了以现代化学键理论为基础的化学结构理论体系。 现代配位化学是既有翔实的实验资料又有坚实的理论基础的完全科学。 ●从定性描述向定量化方向发展 现代配位化学特别是结构配位化学已普遍应用线性代数、群论、矢量分析、拓扑学、数学物理等现代的数学理论和方法了,并且应用电子计算机进行科学 计算,对许多反映结构信息及物理化学性能的物理量进行数学处理。这种数学 计算又与高灵敏度、高精确度和多功能的定量实验测定方法相结合,使对配位 化合物性质和结构的研究达到了精确定量的水平。 ●既分化又综合,出现许多边缘学科 现代配位化学一方面是加速分化,另一方面却又是各分支学科之间的相互 综合、相互渗透,形成了许多新兴的边缘学科。 配位化学的地位 一、现代配位化学的研究领域 现代配位化学主要有七大活跃领域部分,分别为超分子化学、兀酸配休及小分子配体络合物、过渡金属有机络合物、金属原子簇络合物、络合催化、生物配位化学、富勒烯化学-老元素新发现(纳米材料)。 (一)超分子化学 超分子化学是研究两种以上的化学物种通过分子间力相互作用缔结而成为具有特定结构和功能的超分子体系的科学。简而言之,超分子化学是研究多个分子通过非共价键作用,而形成的功能体系的科学。 超分子化学是一门处于化学学科与物理、生命科学相互交叉的前沿学科。它的发展不仅与大环化学(冠醚、穴醚、环糊精、杯芳烃、富勒烯等)的发展密切相关,而且与分子自组装、分子器件和新颖有机材料的研究息息相关。从某种意义上讲,超分子化学将四大基础化学(有机化学、无机化学、分析化学和物理化学)有机地融合成一个整体。 1.分子识别 所谓分子识别是指主体(受体)对客体(底物)选择性结合并产生某种特定功能的过程,是分子组装及超分子功能的基础(锁与钥匙的关系)。 实验三 配合物的生成、性质和应用 一、实验目的 1.了解配合物的生成和组成。 2.了解配合物与简单化合物合复盐的区别。 3.了解配位平衡及其影响因素。 4.了解螯合物的形成条件及稳定性。 5.熟悉过滤盒试管的使用等基本操作。 二、实验原理 由中心离子(或原子)与配体按一定组成和空间构型以配位键结合所形成的化合物称配合物。配位反应是分步进行的可逆反应,每一步反应都存在着配位平衡。 M + nR MR n s n [M R n ] [M ][R ] K 配合物的稳定性可由K 稳 (即K s )表示,数值越大配合物越稳定。增加配体(R)或金属离子(M)浓度有利于配合物(MRn)的形成,而降低配体和金属离子的浓度则有利于配合物的解离。如溶液酸碱性的改变,可能引起配体的酸效应或金属离子的水解等,就会导致配合物的解离;若有沉淀剂能与中心离子形成沉淀的反应发生,引起中心离子浓度的减少,也会使配位平衡朝离解的方向移动;若加入另一种配体,能与中心离子形成稳定性更好的配合物,则同样导致配合物的稳定性降低。若沉淀平衡中有配位反应发生,则有利于沉淀溶解。配位平衡与沉淀平衡的关系总是朝着生成更难解离或更难溶解物质的方向移动。 配位反应应用广泛,如利用金属离子生成配离子后的颜色、溶解度、氧化还原性等一系列性质的改变,进行离子鉴定、干扰离子的掩蔽反应等。 三、仪器和试剂 仪器:试管、离心试管、漏斗、离心机、酒精灯、白瓷点滴板。 试药:H 2SO 4 (2mol·L -1)、HCl (1mol·L -1)、NH 3·H 2O (2, 6mol·L -1)、NaOH (0.1, 2mol·L -1) 、 CuSO 4 (0.1mol·L -1, 固体)、HgCl 2 (0.1mol·L -1)、KI (0.1mol·L -1)、BaCl 2 (0.1mol·L -1 )、K 3Fe (CN)6 (0.1mol·L -1)、NH 4Fe (SO 4)2 (0.1mol·L -1)、FeCl 3 (0.1mol·L -1 )、KSCN (0.1mol·L -1)、NH 4F (2mol·L -1)、(NH 4)2C 2O 4 (饱和)、AgNO 3 (0.1mol·L -1)、NaCl (0.1mol·L -1)、KBr (0.1mol·L -1 )、 Na 2S 2O 3 (0.1mol·L -1,饱和)、Na 2S (0.1mol·L -1)、FeSO 4 (0.1mol·L -1)、NiSO 4 (0.1mol·L -1) 、CoCl 2 (0.1mol·L -1)、CrCl 3 (0.1mol·L -1)、EDTA (0.1mol·L -1 )、乙醇 (95%)、CCl 4、邻菲罗啉 (0.25%)、二乙酰二肟(1%)、乙醚、丙酮。 四、实验内容 1.配合物的生成和组成 (1)配合物的生成 在试管中加入0.5g CuSO 4·5H 2O (s), 加少许蒸馏水搅拌溶解,再逐滴加入2mol·L -1的氨水溶液,观察现象,继续滴加氨水至沉淀溶解而形成深蓝色溶液,然后加入2mL 95%乙醇,振荡试管,有何现象?静置2分钟,过滤,分出晶体。在滤纸上逐滴加入2 mol·L -1NH 3·H 2O 溶液使晶体溶解,在漏斗下端放一支试管承接此溶液,保留备用。写出相应离子方程式。 现象:有浅蓝色沉淀碱式硫酸铜生成:Cu 2++ 2NH 3·H 2O=Cu 2(OH)2SO 4+2NH 4+ 继续滴加沉淀溶解加入乙醇,现象和解释: 析出C u(N H 3)4 S O 4(蓝色) (2)配合物的组成 将上述溶液分成2份,在一支试管中滴入2滴0.1mol·L -1BaCl 2溶液,另一支试管滴入2滴0.1mol·L -1NaOH 溶液,观察现象,写出离子方程式。 配位聚合物在光电磁材料中的应用 摘要:配位聚合物由于其特殊的结构及其在光电磁等方面优异的性能引起了科学家的广泛关注。本文综述了金属有机化合物在光电磁材料中的应用,并对新型多功能材料在设计、合 成与应用方面的广阔前景作了展望。 关键词:配位聚合物;多功能材料;非线性光学;材料化学 引言: 配位聚合物(coordination polymers)或金属-有机框架(metal-organic frameworks,简称 MOFs)是指利用金属离子与有机桥联配体通过配位键合作用而形成的一类具有一维,二维或三维无限网络结构的配位化合物[1]。近年来,配位聚合物作为一种新型的功能化分子材料以其良好的结构可裁性和易功能化的特性引起了研究者浓厚的兴趣。配合物有无机的金属离子和有机配体,因此它兼有无机和有机化合物的特性,而且还有可能出现无机化合物和有机化合物均没有的新性质。配位聚合物分子材料的设计合成、结构及性能研究是近年来十分活跃的研究领域之一,它跨越了无机化学、配位化学、有机化学、物理化学、超分子化学、材料化学、生物化学、晶体工程学和拓扑学等多个学科领域,它的研究对于发展合成化学、结构化学和材料化学的基本概念及基础理论具有重要的学术意义,同时对开发新型高性能的功能分子材料具有重要的应用价值[2-7]。并对分子器件和分子机器的发展起着至关重要的作用。配位聚合物在新的分子材料中将发挥重要的作用。配位化学理论在材料的分子设计中也将起着重要的指导作用。 材料按其性能特征和用途大致可划分为结构材料和功能材料两大类。功能材料种类繁多,功能各异,其共同的特点和发展趋势是:(1) 性能优异;(2) 分子化;(3) 巨大的应用前景。金属有机光电磁材料综合了这几方面特点,将发展成为新一代材料,其结构和性能决定了它的应用越来越广泛。以下是金属有机化合物分别在光电磁材料中的应用。 1配位聚合物在光学材料中的应用 配位聚合物的光学性质研究主要集中在光致发光、电致发光以及非线性光学等方面 配位化学进展 配位化学是在无机化学基础上发展起来的一门边沿学科。它所研究的主要对象为配位化合物(CoordinationCompounds,简称配合物)。早期的配位化学集中在研究以金属阳离子受体为中心(作为酸)和以含N、O、S、P等给体原子的配体(作为碱)而形成的所谓"Werner配合物"。第二次世界大战期间,无机化学家在围绕耕耘周期表中某些元素化合物的合成中得到发展,在工业上,美国实行原子核裂变曼哈顿(Manhattan)工程基础上所发展的铀和超铀元素溶液配合物的研究。以及在学科上,195l年Panson和Miler对二茂铁的合成打破了传统无机和有机化合物的界限。从而开始了无机化学的复兴。 当代的配位化学沿着广度、深度和应用三个方向发展。在深度上表现在有众多与配位化学有关的学者获得了诺贝尔奖,如Werner创建了配位化学,Ziegler和Natta的金属烯烃催化剂,Eigen的快速反应。Lipscomb的硼烷理论,Wnkinson 和Fischer发展的有机金属化学,Hoffmann的等瓣理论Taube研究配合物和固氮反应机理,Cram,Lehn和Pedersen 在超分子化学方面的贡献,Marcus的电子传递过程。在以他们为代表的开创性成就的基础上,配位化学在其合成、结构、性质和理论的研究方面取得了一系列进展。在广度上表现在自Werner创立配位化学以来,配位化学处于无机化学趼究 的主流,配位化合物还以其花样繁多的价键形式和空间结构在化学理论发展中。及其与其它学科的相互渗透中。而成为众多学科的交叉点。在应用方面,结合生产实践。配合物的传统应用继续得到发展。例如金属簇合物作为均相催化剂,在能源开发中C1化学和烯烃等小分子的活化,螯合物稳定性差异在湿法冶金和元素分析、分离中的应用等。随着高新技术的日益发展。具有特殊物理、化学和生物化学功能的所谓功能配合物在国际上得到蓬勃的发展。 自从Werner创建配位化学至今100年以来,以Lehn为代表的学者所倡导的超分子化学将成为今后配位化学发展的另一个主要领域。人们熟知的化学主要是研究以共价键相结合的分子的合成、结构、性质和变换规律。超分于化学可定义为分子间弱相互作用和分子组装的化学。分子间的相互作用形成各种化学、物理和生物中高选怿性的识别、反应、传递和调制过程。而这些过程就导致超分子的光电功能和分子器件的发展。 我国配位化学的研究在中华人民共和国成立前几乎属于空白。1949年后随着国家经济建设的发展,仅在个别重点高等院校及科研单位开展了这方面的教学和科研工作,60年代中期以前。主要工作集中在简单配合物的合成、性质、结构及其应用方面的研究。特别是在溶液配合物的平衡理论、混合和多核配合物的稳定性、取代动力学、过渡金属配位催化以 配位化学论文 分子轨道理论的发展及其应用 160113004 2013级化教一班马慧敏 一、前言 价建理论、分子轨道理论和配位场理论是三种重要的化学键理论。三、四十年代,价键理论占主要的地位。五十年代以来由于分子轨道理论容易计算且得到实验(光电能谱)的支持,取得了巨大的发展,逐渐占优势。价建理论不但在理论化学上有重要的意义(下文中将详细介绍)。在应用领域也有重要的发展,如分子轨道理论计算有机化合物的吸收光谱用于染料化学;前线分子轨道理论在选矿中的研究等等。 二、简介 1、分子轨道理论产生和发展 在分子轨道理论出现以前,价键理论着眼于成键原子间最外层轨道中未成对的电子在形成化学键时的贡献,能成功地解释了共价分子的空间构型,因而得到了广泛的应用。但如能考虑成键原子的层电子在成键时贡献,显然更符合成键的实际情况。1932年,美国化学家 Mulliken RS和德国化学家HundF 提出了一种新的共价键理论——分子轨道理论(molecular orbital theory),即MO法。该理论注意了分子的整体性,因此较好地说明了多原子分子的结构。目前,该理论在现代共价键理论中占有很重要的地位。 以下是各个年代提出的关于分子轨道理论的一些重要理论和方法,是分子轨道理论发展过程中的几个里程碑! 1926-1932年,在讨论分子光谱时,Mulliken和Hund提出了分子轨道理论。 认为:电子是在整个分子轨道中运动,不是定域化的。他们还提出能级图、成键、反键轨道等重要的概念。 1931-1933年,Hukel提出了一种简单的分子轨道理论,用于讨论共轭分子的性质,相当成功。 1950年,Boys用Guass函数研究原子轨道,解决了多中心积分问题,是今天广为利用的自洽场分子轨道理论的基础,在量子化学的研究中占有重要地位。 1951年,Roothaan在Hartree-Fock方程的基础上,把分子轨道写成原子轨道的线性组合,得到Roothaan方程。 1952年,福井谦一提出前线分子轨道理论,用以讨论分子的化学活性和分子间相互作用等,可以解释许多实验结果。 1965年,Woodward和Hoffman提出分子轨道对称守恒原理,发展成讨论基元反应发生可能性的重要规则。用于指导某些复杂化合物分子的合成。 2、分子轨道理论的含义和一些重要分子轨道的构成方法 1)分子轨道理论的含义 目录 摘要 (2) 关键词 (2) Abstract (2) Keywords (2) 引言 (2) 1.配位键,配位数 (2) 1.1 配位键 (2) 1.1.1 σ键 (2) 1.1.2 П键 (3) 1.2 配位数 (3) 2.轨道杂化与配合物的空间构型 (3) 2.1轨道杂化 (3) 2.2 配合物的内、外轨型 (4) 2.2.1 配为单元的形成 (4) 2.2.2 应用举例 (4) 2.3 中心离子的杂化轨道类型与配离子的空间构型 (5) 3.价键理论的应用及不足 (5) 3.1 价键理论的应用 (5) 3.2 价键理论的不足 (6) 4.结束语 (6) 参考文献 (6) 配合物的价键理论 摘要:价键理论是由Pauling将杂化轨道理论应用于配合物而得到的,20世纪30年代到40年代主要用于讨论配合物的化学键。价键理论可以解释配合物的结构、稳定性、磁性等现象,但它对配合物的光谱等不能作出解释。[1] 关键词:配合物;配位键;价键理论。 Abstract:The valence bond theory is put forward by Pauling,and he applied the complex theory to Coorination Compounds.During the 30th and 40th of 20 century,it is always used to discuss the configurational field of Coorination compounds.It can be used to research the structure of complexes,magnetism and stability but the properties of spectrum. Keywords:complex compound; Valence bond theory。 引言: 配合物价键理论、晶体场理论、分子轨道理论和配位场理论是研究配合物结构与性质的4个著名理论[2]特别是上世纪30年代初Pauling提出的价键理论及1923-1935年Bethe 和Hvan Vlecer提出的晶体场理论。由于这两个理论简明清晰,使用方便,因此在研究配合物结构与性质时得到广泛应用。而配合物的分子轨道理论内容复杂,计算繁难,使用不便,特别是目前还没有简便实用的各种几何构型配合物的分子轨道能级图,因此分子轨道理论的应用受到了限制。[3]目前使用广泛的价键理论和晶体场理论本身也存在着明显的缺陷。[4]本文就价键理论的内容、应用及不足略做介绍。 1.配位键,配位数 1.1 配位键 配合物的价键理论认为,配体提供来形成键的电子是进入中心原子的原子轨道的,或者说,只有中心原子提供原子轨道来接受配体提供的电子对才能形成配位键,有σ键和П键。 1.1.1 σ键 配位化学的现状及发展 专业班级:化学(师范类)一班姓名:刘楠楠课程名称:配位化学 摘要:配位化学已成为当代化学的前沿领域之一。它的发展打破了传统的有机化学和无机化学之间的界线。其新奇的特殊性能在生产实际中得到了重大的应用,花样繁多的价健理论及空间结构引起了结构化学和理论化学家的深切关注。它和物理化学、有机化学、生物化学、固体化学、环境化学相互渗透,使其成为贯通众多学科的交叉点。本文将介绍配位化学在近几年的现状和发展。 关键词:配位化学;现状;发展 配位化学是在无机化学基础上发展起来的一门交叉学科,50年代以来配位化学以其与有机合成化学和结构化学相结合为特点,开始了无机化学的复兴时期,从而在实际上打破了传统的无机、有机和物理化学间的界限,进而成为各化学分支的结合点。配合物以其花样繁多的价键和空间结构促进了基础化学的发展,又以其特殊的性质在生产实践和科学实验中取得了重大的应用。配位化学是化学学科中最活跃的,具有很多生长点的前沿学科之一,它的近期发展趋势如下。 1.具有特殊性质和特殊结构配合物的合成、结构及性能的研究 各种大环、夹心、多核、簇状、非常氧化态、非常配位数、混合价态及各种罕见构型配合物的合成、结构、热力学、动力学和反应性的研究正在深入。其中巨型原子簇的研究已成为阐明金属原子化学和固体金属化学异同的桥梁;新型球型大环,聚邻苯酚脂大环配体对某些金属离子具有特殊高的选择性;在CO,CO2,H2和CH4等小分子配合物及活化方面,已发现用Co+,Li+双核配合物不仅可与CO2配位,并使其活化,而形成C—C键;此外H2的配合物研究及H2的活化亦在深入。 配合物合成、结构和性能研究方面,近年来的一个引人注目的动向是配位化学和固体化学的交叉[1]。一系列具有链状、层片状和层柱状特殊结构的配合物已经合成。对它们的性质和结构,正在进行系统研究。 2.溶液配位化学研究 溶液配位化学研究正在继续深入,但已具有新的内容。在取代反应动力学及机理方面,近年工作集中在金属碳基配合物的研究上。已知配体的空间效应,电配位化学基础
现代配位化学研究的领域及配位学的应用
实验三_配合物的生成、性质与应用
配位化学
配位化学进展
配位化学论文设计---分子轨道理论
配合物的价键理论
配位化学现状及发展
相关主题
文本预览