
自旋轨道耦合计算过程探索
1.经验总结
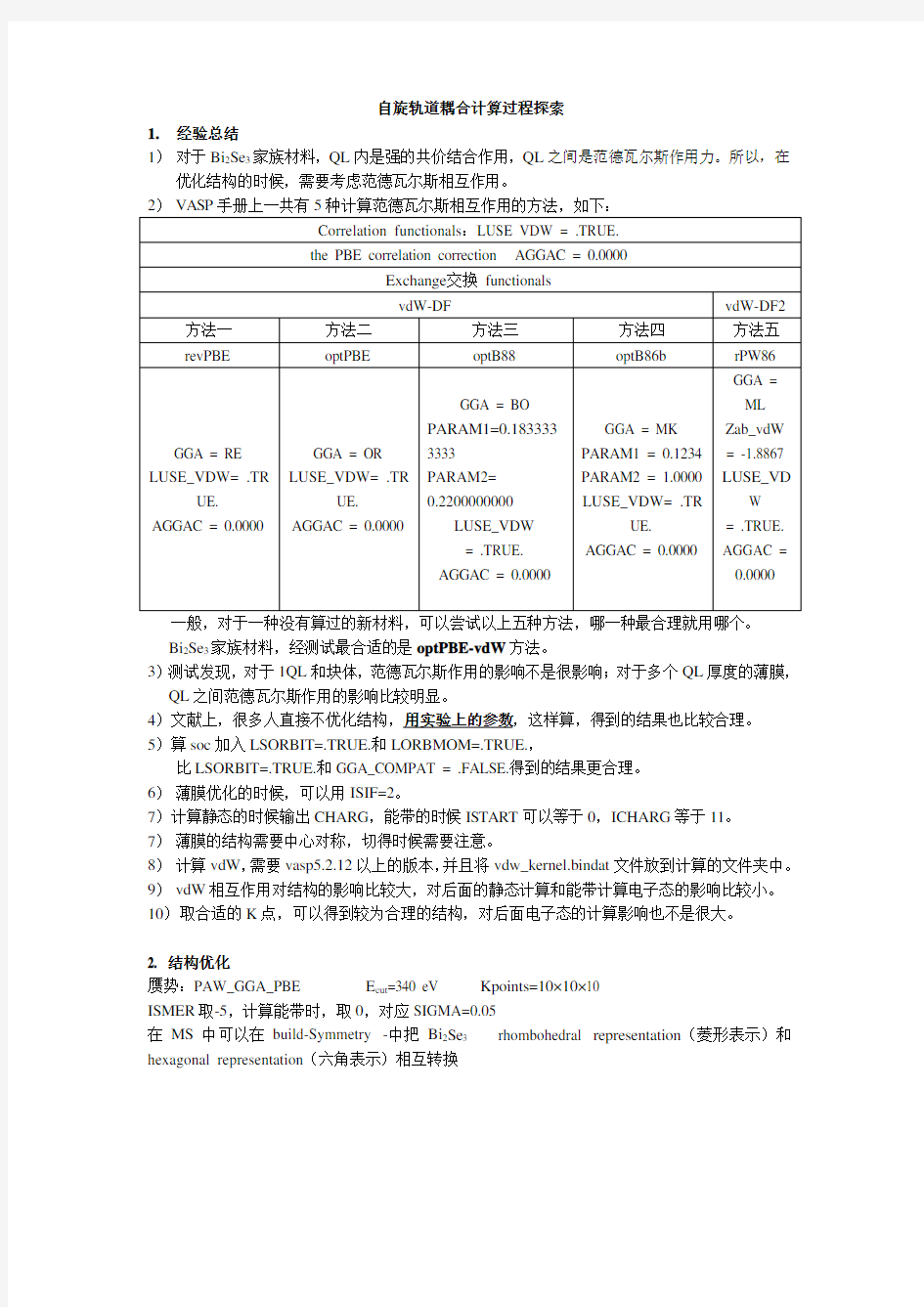
1)对于Bi2Se3家族材料,QL内是强的共价结合作用,QL之间是范德瓦尔斯作用力。所以,在优化结构的时候,需要考虑范德瓦尔斯相互作用。
一般,对于一种没有算过的新材料,可以尝试以上五种方法,哪一种最合理就用哪个。
Bi2Se3家族材料,经测试最合适的是optPBE-vdW方法。
3)测试发现,对于1QL和块体,范德瓦尔斯作用的影响不是很影响;对于多个QL厚度的薄膜,QL之间范德瓦尔斯作用的影响比较明显。
5)算soc加入LSORBIT=.TRUE.和LORBMOM=.TRUE.,
比LSORBIT=.TRUE.和GGA_COMPAT = .FALSE.得到的结果更合理。
6)薄膜优化的时候,可以用ISIF=2。
7)计算静态的时候输出CHARG,能带的时候ISTART可以等于0,ICHARG等于11。
7)薄膜的结构需要中心对称,切得时候需要注意。
8)计算vdW,需要vasp5.2.12以上的版本,并且将vdw_kernel.bindat文件放到计算的文件夹中。9)vdW相互作用对结构的影响比较大,对后面的静态计算和能带计算电子态的影响比较小。10)取合适的K点,可以得到较为合理的结构,对后面电子态的计算影响也不是很大。
2. 结构优化
赝势:PAW_GGA_PBE E cut=340 eV Kpoints=10×10×10
ISMER取-5,计算能带时,取0,对应SIGMA=0.05
在MS中可以在build-Symmetry -中把Bi2Se3 rhombohedral representation(菱形表示)和hexagonal representation(六角表示)相互转换
图中黑色t 1、t 2、t 3基矢围成菱形原胞,用于计算块体,红色方框包含一个五元层 计算能带的布里渊区高对称点:
块体:文献中倒空间高对称点坐标Г(0 0 0)-Z(π π π)-F(π π 0)-Г(0 0 0)-L(π 0 0), 根据正空间和倒空间坐标的转换关系,得到正空间中高对称点的坐标: Г(0 0 0)-Z(0.5 0.5 0.5)-F(0.5 0.5 0)-Г(0 0 0)-L(0 0 -0.5) KPOINTS 20 Line-mode Rec
0.0 0.0 0.0 !Г 0.5 0.5 0.5 ! Z 0.5 0.5 0.5 ! Z 0.5 0.5 0.0 ! F 0.5 0.5 0.0 ! F 0.0 0.0 0.0 !Г 0.0 0.0 0.0 !Г 0.0 0.0 -0.5 ! L
[通过比较结构,发现Ecut=580,KPOINTS=151515,得到的结构比较靠谱]
3. 块体soc 的计算 文献能带结构图:
块体(Bi 2Se 3-VASP-GGA-PAW-PBE )
我们的结果(未考虑vdW+静态和能带都加soc计算结果与文献基本符合):
4.薄膜的计算
薄膜:Kpoints=10×10×1
计算能带的K点和石墨烯(六角晶胞的)的K点一样:
KPOINTS
20
Lone-mode
Rec
0.66666667 0.33333333 0.0 !K
0.0 0.0 0.0 !Г
0.0 0.0 0.0 !Г
0.5 0.0 0.0 !M
考虑薄膜的对称性
由MS六角结构,沿(001)方向切割,可以得到两种以Se原子作为表面原子的薄膜,如下图,分别为1QL和3QL的两种切法,右图比左图对称性要更好一些,这一区别在计算过程中会导致巨大的区别,我们通过比较,发现,只有右图的结果,才可以得到合理的结果,尤其是在多个QL的情况。
用左边结构得到的结果(Bi 2Se 3):
用左边结构得到的结果(Bi 2Te 3):
用右边结构得到的结果(Bi2Se3):
1QL—根据块体的数据得到薄膜,分以下两种情况计算:
1.不优化结构,scf不加soc,bands加soc
2.用块体的参数,加vdW优化结构,scf不加soc,bands加soc
1QL—在静态中也加入soc
1.不优化结构,scf和bands加入LORBMOM=.TRUE.,LSORBIT=.TRUE.
2.不优化结构,scf和bands加入LORBMOM=.TRUE.,GGA_COMPA T=.FALSE.
3.优化结构,scf和bands加入LORBMOM=.TRUE.,LSORBIT=.TRUE.
4.优化结构,scf和bands加入LORBMOM=.TRUE.,GGA_COMPA T=.FALSE.
文献结果:
上图是没有进行离子弛豫的1QL ~6QL的Bi2Se3薄膜能带结构
上图采用optPBE-vdW 泛函进行离子弛豫1QL~6QL的Bi2Se3薄膜能带结构
上图是实验观测的1QL ~6QL(12356)的Bi2Se3薄膜能带结构.
5.调试过程错误总结
错误1:VERY BAD NEWS! Internal error in subroutine IBZKPT: Reciprocal lattice and k-lattice belong todifferent class of lattices. Often results are still useful (48)
Internal内部 subroutine子程序 Reciprocal倒数的
非常严重的错误!子程序IBZKPT中内部错误:倒格子和k点网格属于不同类型的格子。通常结果还是有用的。
解决方案:根据所用集群,修改INCAR中NPAR。将NPAR=4变成NPAR=1,已解决!
错误2:
internal ERRORRSPHER:running out of buffer 0 0
13 1 0
nonlr.F:Out of bufferRSPHER
解决方案:根据CPU的数量,修改INCAR中NPAR,将NPAR=1修改成4(或者2),问题得以解决。
错误3:WARNING: Sub-Space-Matrix is not hermitian in DAV 4 -4.681828688433112E-002 Sub-Space-Matrix 子空间矩阵、亚空间矩阵 Hermitian厄米共轭
警告:戴维森方法(DAV)中的子空间矩阵不是厄米共轭的。
解决方案:只需调整 AMIX, BMIX的值,把他们设置小一些。一般采用其默认值,除非在电子迭代难以收敛的情况,才手动设置AMIX和BMIX等参数值。经对Mixing方法的调试,通过将默认AMIX=0.4,修改成AMIX=0.2(或0.3),问题得以解决。
Mixing方法:
IMIX=type ofmixing混合、混频, AMIX=linear mixing parameter, AMIN=minimal mixing parameter,
BMIX=cutoffwave vector for Kerker mixing scheme, AMIX_MAG=linear mixing parameter for magnetization,
BMIX_MAG=cutoffwave vector for Kerker mixing scheme for mag, WC=weight factor for each step inBroyden mixing scheme,
INIMIX=type ofinitial for each step in Broyden mixing scheme, MIXPRE=type of preconditioning in Broyden mixing scheme,
MAXMIX=maximumnumber steps stored in Broyden mixer.
错误4:WARNING: Sub-Space-Matrix is not hermitian in DAV 1 -7.626640664998020E-003 解决方案:在INCAR中加上IALG=Fast 已解决!(1QL、2QL已解决,3QL以上未解决)IALG=Fast (两种方法混用)
IALGO=38 IALG=Normal 电子优化采用blockedDavidson方法
IALGO=48 IALG=Very_Fast 电子优化采用RMM-DIIS算法
错误5:ADVICE TO THIS USER RUNNING 'VASP/VAMP' (HEAR YOUR MASTER'S
VOICE ...): You have a(more or less) 'small supercell' and for smaller cells it is recommended to usethe reciprocal-space projection scheme! The real space optimization is notefficient for small cells and it is also less accurate ... Therefore setLREAL=.FALSE. in the INCAR file
解决方案:对于较小的晶胞(原子数小于20),设置LREAL=.FALSE.,计算结果比较精确。而对于较大的晶胞,设置LREAL=Auto,这样计算速度比较快。
对于1QL 2QL 3QL原子数分别为5、10、15,LREAL=.False.
对于4QL 5QL 6QL原子数分别为20、25、30,LREAL=Auto
错误6:自旋轨道耦合计算时,静态和能带计算中出现的错误:
ERROR: non collinear calculations require that VASP is compiled withoutthe flag -DNGXhalf and
-DNGZhalf
错误:非线性计算需要编译过的VASP,VASP中不包含-DNGXhalf 和-DNGZhalf
解决方案:重新编译VASP。
don't forget that you may have to re-compile vasp without any of theprecompiler (CPP) flags set:
-DNGXhalf, -DNGZhalf, -DwNGXhalf, -DwNGZhalf , asnecessary for non-collinear runs in general for non-collinear magnetism
不要忘记如果你用的vasp不包含任何预编译程序命令 -DNGXhalf, -DNGZhalf, -DwNGXhalf,
-DwNGZhalf ,你必须重新编译vasp只有编译过,因为这些参数通常对于非线性磁性计算是必要的.
错误7: SB-3QL计算总是出现这个错误 VERY BAD NEWS! internal error in subroutine SGRCON: Found some non-integer element in rotation matrix 3
解决办法:首先检查POSCAR是否有问题。MS重新获取POSCAR,参与计算,已解决。
错误8: WARNING: random wavefunctions but no delay for mixing, default for NELMDL
建议解决办法:一般不是很影响,可以继续算。
可以设置NELMDL=-5或者其它的数,是前5步保持电荷密度不变,只弛豫波函数。
错误9: 计算结构得到一个空的,然后算静态也没有数据,能带也没有数据,但都算完了。
输出文件中提示:
mpirun has exited due to process rank 21 with PID 8455 on node node14 exiting improperly. There are two reasons this could occur:1. this process did not call "init" before exiting, but others in the job did. This can cause a job to hang indefinitely while it waits for all processes to call "init". By rule, if one process calls "init", then ALL processes must call "init" prior to termination.2. this process called "init", but exited without calling "finalize". By rule, all processes that call "init" MUST call "finalize" prior to exiting or it will be considered an "abnormal termination"This may have caused other processes in the application to be terminated by signals sent by mpirun (as reported here).
解决方案:这是VASP版本的问题,考虑vdW作用,有的版本的vasp算不了,一般5.2.12以上的版本可以,换一个版本就可以计算。
错误10:计算总是卡住,输出的log文件中只有前面的描述,没有后面的数据,比如:
running on 32 total cores[2016-05-19 18:42:45] distrk: each k-point on 32 cores, 1 groups[2016-05-19 18:42:45] distr: one band on 8 cores, 4 groups[2016-05-19 18:42:45] using from now: INCAR[2016-05-19 18:42:45] vasp.5.3.3 18Dez12 (build May 19 2015 15:36:57) complex[2016-05-19 18:42:45] [2016-05-19 18:42:45] POSCAR found type information on POSCAR Se Bi[2016-05-19 18:42:45] POSCAR found : 2 types and 15 ions[2016-05-19 18:42:45] LDA part: xc-table for Pade appr. of Perdew[2016-05-19 18:42:45] WARNING: stress and forces are not correct[2016-05-19 18:42:46] POSCAR, INCAR and KPOINTS ok, starting setup[2016-05-19 18:42:46] FFT: planning ...[2016-05-19 18:42:47] WAVECAR not read[2016-05-19 18:42:48] charge-density read from file: Bi2Se3scf[2016-05-19 18:42:49] magnetization density of overlapping atoms calculated
解决方案:这种情况一般是由于计算量过大,算不动导致的,通过减少精度,就可以解决。比如,减少K点数量(主要的影响因素)。
错误11:VERY BAD NEWS! internal error in subroutine SGRCON:
Found some non-integer element in rotation matrix 2
解决问题:一般是结构的问题。重新得到POSCAR,可以解决。
Bi 2Se 3自旋轨道耦合性质的计算 一、模型和基本参数: 图(a )黑色t 1、t 2、t 3基矢围成Bi 2Se 3菱形原胞,用于计算块体,红色方框包含一个五元层,是构成薄膜的一个QL 。 计算能带的布里渊区高对称点:Г(0 0 0)-Z(π π π)-F(π π 0)-Г(0 0 0)-L(π 0 0), 根据正空间和倒空间坐标的转换关系, 得到正空间中高对称点的坐标:Г(0 0 0)-Z(0.5 0.5 0.5)-F(0.5 0.5 0)-Г(0 0 0)-L(0 0 -0.5) 空间群: 166号~ R-3M (MS ) ) 3(5 3m R D d (文献) 结构分为:六角晶胞和菱形原胞(Rhombohedral )两种形式 六角晶胞(hexagon):含三个五元层,15个原子 菱形原胞(Rhombohedral ):含5个原子 晶格参数t=9.841, α=24.275 原子坐标: 弛豫值 实验值 Bi(2c) (0.400,0.400,0.400) Bi(2c) (0.398, 0.398, 0.398) Se(1a) (0,0,0) Se(1a) (0,0,0) Se(2c) (0.210, 0.210, 0.210) Se(2c) (0.216, 0.216, 0.216) 赝势:PAW_GGA_PBE E cut =340 eV 块体:Kpoints=11×11×11 薄膜:Kpoints=11×11×1 块体结构优化时,发现Ecut=580,KPOINTS=151515,得到的结构比较合理 计算薄膜真空层统一: 15 ?
ISMER取-5(或取0,对应SIGMA=0.05) 二、计算过程描述: 1)范德瓦尔斯作用力的影响。 手册中一共有5种方法: Correlation functionals:LUSE VDW = .TRUE. the PBE correlation correction AGGAC = 0.0000 Exchange交换functionals vdW-DF vdW-DF2 方法一方法二方法三方法四方法五revPBE optPBE optB88 optB86b rPW86 GGA = RE LUSE_VDW = .TRUE. AGGAC = 0.0000 GGA = OR LUSE_VDW = .TRUE. AGGAC = 0.0000 GGA = BO PARAM1 = 0.1833333333 PARAM2 = 0.2200000000 LUSE_VDW = .TRUE. AGGAC = 0.0000 GGA = MK PARAM1 = 0.1234 PARAM2 = 1.0000 LUSE_VDW = .TRUE. AGGAC = 0.0000 GGA = ML Zab_vdW = -1.8867 LUSE_VDW = .TRUE. AGGAC = 0.0000 经测试,发现方法二optimized Perdew-Burke-Ernzerhof-vdW (optPBE-vdW)是最合适的。并通过比较发现,范德瓦尔斯作用力对块体和单个QL厚度的薄膜的影响很小,对多个QL 厚度的薄膜结构影响比较大,所以优化时需要考虑QL之间的vdW相互作用,而范德瓦尔斯作用力对电子态的影响也比较小,所以,计算静态和能带的时候,可以不考虑。 此外,以往文献中的计算,有的直接采用实验给出的结构参数建模,不再弛豫,计算静态和能带,得到的结果也比较合理。 所以,我们对薄膜采用不优化结构和用optPBE方法优化结构,两种方式。 2)算SOC。 计算材料的自旋轨道耦合性质,一般在优化好的结构基础上,在静态和能带计算是加入特定参数来实现。一般,分两种方式: 第一种是从静态开始,就进行非线性的计算,能带也进行非线性自旋轨道耦合计算。 第二种,则是,在静态时进行非线性计算(按照一般的静态计算进行),产生CHGCAR、WA VECAR,进行能带非线性自旋轨道计算时,读入这两个参数。 V ASP手册推荐使用第二种。 我们通过多次比较发现,使用第一种方法,可以得到更为合理的结果。 3)关于d电子的考虑。 我们分别考虑了Bi原子的两种电子组态: 第一种,含有15个价电子,包含d电子,电子组态5d106s26p3; 第二种,含有5个价电子,不含d电子,电子组态是6s26p3。 通过比较计算结果,发现并没有明显的区别,所有我们选用第二种。
VASP 自旋轨道耦合计算 已有4532 次阅读2011-9-13 20:37|个人分类:VASP|系统分类:科研笔记 将VASP 的makefile 文件中的 CPP 选项中的 -DNGXhalf, -DNGZhalf, -DwNGXhalf, -DwNGZhalf 这4个选项去掉重新编译VASP才能计算自旋轨道耦合效应。 以下是从VASP在线说明书整理出来的非线性磁矩和自旋轨道耦合的计算说明。 非线性磁矩计算: 1)计算非磁性基态产生WAVECAR和CHGCAR文件。 2)然后INCAR中加上 ISPIN=2 ICHARG=1 或 11 !读取WAVECAR和CHGCAR文件 LNONCOLLINEAR=.TRUE. MAGMOM= 注意:①对于非线性磁矩计算,要在x, y 和 z方向分别加上磁矩,如 MAGMOM = 1 0 0 0 1 0 !表示第一个原子在x方向,第二个原子的y方向有磁矩 ②在任何时候,指定MAGMOM值的前提是ICHARG=2(没有WAVECAR和CHGCAR文件)或者ICHARG=1 或11(有WAVECAR和CHGCAR文件),但是前一步的计算是非磁性的(ISPIN=1)。 磁各向异性能(自旋轨道耦合)计算:
注意: LSORBIT=.TRUE. 会自动打开LNONCOLLINEAR= .TRUE.选项,且自旋轨道计算只适用于PAW赝势,不适于超软赝势。 自旋轨道耦合效应就意味着能量对磁矩的方向存在依赖,即存在磁各向异性能(MAE),所以要定义初始磁矩的方向。如下: LSORBIT = .TRUE. SAXIS = s_x s_y s_z (quantisation axis for spin) 默认值: SAXIS=(0+,0,1),即x方向有正的无限小的磁矩,Z方向有磁矩。 要使初始的磁矩方向平行于选定方向,有以下两种方法: MAGMOM = x y z ! local magnetic moment in x,y,z SAXIS = 0 0 1 ! quantisation axis parallel to z or MAGMOM = 0 0 total_magnetic_moment ! local magnetic moment parallel to SAXIS (注意每个原子分别指定) SAXIS = x y z !quantisation axis parallel to vector (x,y,z),如 0 0 1 两种方法原则上应该是等价的,但是实际上第二种方法更精确。第二种方法允许读取已存在的WAVECAR(来自线性或者非磁性计算)文件,并且继续另一个自旋方向的计算(改变SAXIS 值而MAGMOM保持不变)。当读取一个非线性磁矩计算的WAVECAR时,自旋方向会指定平行于SAXIS。 计算磁各向异性的推荐步骤是: 1)首先计算线性磁矩以产生WAVECAR 和CHGCAR文件(注意加入LMAXMIX)。 2)然后INCAR中加入: LSORBIT = .TRUE. ICHARG = 11 ! non selfconsistent run, read CHGCAR
自旋轨道耦合计算过程探索 1.经验总结 1)对于Bi2Se3家族材料,QL内是强的共价结合作用,QL之间是范德瓦尔斯作用力。所以,在优化结构的时候,需要考虑范德瓦尔斯相互作用。 一般,对于一种没有算过的新材料,可以尝试以上五种方法,哪一种最合理就用哪个。 Bi2Se3家族材料,经测试最合适的是optPBE-vdW方法。 3)测试发现,对于1QL和块体,范德瓦尔斯作用的影响不是很影响;对于多个QL厚度的薄膜,QL之间范德瓦尔斯作用的影响比较明显。 5)算soc加入LSORBIT=.TRUE.和LORBMOM=.TRUE., 比LSORBIT=.TRUE.和GGA_COMPAT = .FALSE.得到的结果更合理。 6)薄膜优化的时候,可以用ISIF=2。 7)计算静态的时候输出CHARG,能带的时候ISTART可以等于0,ICHARG等于11。 7)薄膜的结构需要中心对称,切得时候需要注意。 8)计算vdW,需要vasp5.2.12以上的版本,并且将vdw_kernel.bindat文件放到计算的文件夹中。9)vdW相互作用对结构的影响比较大,对后面的静态计算和能带计算电子态的影响比较小。10)取合适的K点,可以得到较为合理的结构,对后面电子态的计算影响也不是很大。 2. 结构优化 赝势:PAW_GGA_PBE E cut=340 eV Kpoints=10×10×10 ISMER取-5,计算能带时,取0,对应SIGMA=0.05 在MS中可以在build-Symmetry -中把Bi2Se3 rhombohedral representation(菱形表示)和hexagonal representation(六角表示)相互转换
BI YE SHE JI (20 届) 自旋轨道耦合费米气体的研究
自旋轨道耦合费米气体的研究 摘要 本文对于自旋轨道耦合费米气体的研究,是从一维自旋轨道耦合费米气体与三维自旋轨道耦合费米气体这两个方面进行描述的。对一维自旋轨道耦合费米气体的分析是从内容、模型、以及相应处理三个方面进行,主要说明在横向磁场的在光学晶格中的一维自旋轨道耦合的费米气体这个模型,然后运用玻色化的预测进行处理,对自旋轨道耦合相互作用费米气体的相图分析,得出除了一个半填充绝缘相在强电场限制和一个带绝缘体,我们确定一个在弱场中的LE相和在强场中的另一个超导相的结论。在另一方面,对于三维自旋轨道耦合费米气体,我们是在塞曼场的平面内和平面外,对自旋轨道耦合的费米气体超流相进行系统的研究。描述了一些系统所拥有的特征,比如福尔德-弗雷尔配对、马约喇纳费米子、范尔费米子和无间隙的拓扑超流态。 关键词:费米气体、自旋轨道耦合、福尔德-弗雷尔配对、马约喇纳费米子、范尔费米子、拓扑超流态 ABSTRACT The analysis of spin-orbit coupled Fermi gas consists of two aspects, one is the analysis of spin-orbit coupled one dimensional Fermi gas , and another is the analysis of spin-orbit coupled three dimensional Fermi gas. On the one hand, as for the analysis of spin-orbit coupled one dimensional Fermi gas, we investigate it for its introduces, model and research. We discussed that the model of a spin-orbit coupled interacting Fermi gas in the 1D optical lattice with a transverse magnetic field., and then study it by bosonization predition. Studying the phase diagram of a spin-orbit coupled interacting Fermi gas.Finally we conclude that besides a half-filled insulating phase in the strong field limit and a band insulator we identify a LE phase in the weak field and another superconducting phase in the strong field On the other hand, a systematic investigation of the superfluid phases of a spin-orbit coupled Fermi gas with both in-plane and out-of-plane Zeeman fields .There are some characteristics, such as Fulde-Ferrell pairing, Majorana fermions, Weyl fermions and gapless topological superfluidity Key Words: Fermi gas, spin-orbit coupled,Fulde-Ferrell pairing, Majorana fermions, Weyl fermions ,topological superfluidity
目录 摘要.......................................................................................................................................I Abstract...............................................................................................................................II 第1章 绪论.. (1) 1.1自旋电子学 (1) 1.2 磁纳米结构 (5) 1.3 磁纳米结构中电子自旋极化效应 (8) 1.4 硕士学位论文的研究工作 (11) 第2章 研究方法和理论 (13) 2.1 改进的转移矩阵法 (13) 2.2Landauer-Büttiker超微结构电导理论 (16) 2.3 本章小结 (18) 第3章 自旋-轨道耦合调制下磁垒纳米结构中电子自旋极化效应 (19) 3.1 引言 (19) 3.2 模型和公式 (20) 3.3 结果和讨论 (23) 3.4 本章小结 (30) 第4章 自旋-轨道耦合调制下复合磁电垒纳米结构中电子自旋极化效应 (31) 4.1 引言 (31) 4.2 模型和公式 (32) 4.3 结果和讨论 (36) 4.4 本章小结 (42) 第5章 结论与展望 (44) 参考文献 (46) 个人简历、申请学位期间的研究成果及发表的学术论文 (52) 致谢 (53) III 万方数据
车辆-轨道耦合动力学理论在轨道下沉变形 研究中的应用1 高建敏,翟婉明 西南交通大学牵引动力国家重点实验室,四川成都(610031) E-mail:jianmingao04@https://www.doczj.com/doc/c03196413.html, 摘要:提出了将车辆-轨道耦合动力学理论引入轨道下沉变形研究的分析方法。通过将车辆-轨道垂向耦合振动模型和轨道累积下沉计算模型相结合,以轨道结构动力学响应参量和轨面高低不平顺状态变化等作为两者间的联结纽带,从车辆-轨道耦合动力学角度研究了轨道的下沉变形特性。研究结果表明,随着轨道动荷载重复作用次数的增加,轨道下沉量逐渐累积,轨面初始不平顺对轨道下沉变化影响较大。车辆-轨道耦合振动系统和轨道下沉变形处于特定的相互作用过程之中,受轨道累积下沉变形的影响,轮轨力、轨道结构响应加剧。 关键词:车辆;轨道;动力学;累积变形;下沉 中图分类号:U260.11 1. 引言 铁路有碴轨道在运营使用过程中,由于其自身特点,会不可避免地产生残余变形。这种残余变形随着列车荷载的反复作用,逐渐累积,最终导致轨道结构的下沉。轨道累积下沉快慢及下沉量大小直接关系到轨道的维修模式和成本[1]。因此,研究轨道的下沉变形累积特性,预测下沉发展趋势,对经济、合理地安排轨道养护维修,保证列车安全、平稳、不间断运行,具有重要意义。 有关轨道下沉变形的研究最初以试验研究为主,英国、日本、前苏联等国均通过大量试验和现场调查,建立了各自的轨道下沉(主要是道床)计算模型[2~5],我国在道床下沉计算模型方面也有研究,但相对较少[1,6]。近年来,随着计算机技术的大力发展,使大型仿真分析研究成为可能,研究人员开始探索利用计算机仿真技术,通过数值算法,从理论角度深入研究有如轨道下沉这样的复杂问题,代表性国家主要有英国、瑞典和日本[7~9]。国内在轨道下沉仿真分析方面开展的研究甚少,至今尚未看到较为相关的文献资料。因此,本文在国外研究经验基础上,基于车辆-轨道耦合动力学理论和轨道下沉变形法则,通过将车辆-轨道耦合振动系统和轨道下沉变形相联结,开展了有关轨道动态下沉变形特性以及车辆-轨道耦合振动系统与轨道下沉变形间相互影响关系的研究。 2 研究方法及仿真计算模型 2.1 轨道下沉研究方法 铁路运输属轮轨系统运输模式,车辆与轨道系统处于特定的耦合振动形态之中,车辆与轨道相互作用,轨道几何形位的变化,轨道结构的变形和损伤,是车辆系统和轨道系统相互作用再加上外界自然因素的影响而形成的。轨道的下沉变形是由于列车-轨道相互作用产生的轨道动荷载诱发而产生的,而轨道下沉变形结果又会叠加于原始轨道形态之上,进一步影响到车辆与轨道动态作用。可见,轨道的下沉变形和车辆-轨道耦合系统之间是一个相互作用的过程,研究轨道的下沉变形离不开对车辆-轨道耦合振动系统的分析和研 1. 本课题得到教育部创新团队计划资助(IRT0452)、国家博士学科点基金项目(20030613011)和西南交通大学博士创新基金的资助。
VASP自旋轨道耦合计算错误汇总 静态计算时,报错: VERY BAD NEWS!Internal内部error in subroutine子程序IBZKPT: Reciprocal倒数的lattice and k-lattice belong to different class of lattices.Often results are still useful (48) INCAR参数设置: 对策:根据所用集群,修改INCAR中NPAR。将NPAR=4变成NPAR=1,已解决! 错误:sub space matrix类错误 报错:静态和能带计算中出现警告:WARNING:Sub-Space-Matrix is not hermitian共轭in DAV 结构优化出现错误: WARNING:Sub-Space-Matrix is not hermitian in DAV4-4.681828688433112E-002 对策:通过将默认AMIX=0.4,修改成AMIX=0.2(或0.3),问题得以解决。 以下是类似的错误: WARNING:Sub-Space-Matrix is not hermitian in rmm-3.00000000000000 RMM:22-0.167633596124E+02-0.57393E+00-0.44312E-0113260.221E+00BRMIX: very serious problems the old and the new charge density differ old charge density:28.00003new28.060930.111E+00 错误: WARNING:Sub-Space-Matrix is not hermitian in rmm-42.5000000000000 ERROR FEXCP:supplied Exchange-correletion table is too small,maximal index:4794 错误:结构优化Bi2Te3时,log文件: WARNING in EDDIAG:sub space matrix is not hermitian1-0.199E+01 RMM:2000.179366581305E+01-0.10588E-01-0.14220E+007180.261E-01 BRMIX:very serious problems the old and the new charge density differ old charge density:56.00230new124.70394 66F=0.17936658E+01E0=0.18295246E+01d E=0.557217E-02 curvature:0.00expect dE=0.000E+00dE for cont linesearch0.000E+00 ZBRENT:fatal error in bracketing please rerun with smaller EDIFF,or copy CONTCAR to POSCAR and continue 但是,将CONTCAR拷贝成POSCAR,接着算静态没有报错,这样算出来的结果有问题吗? 对策1:用这个CONTCAR拷贝成POSCAR重新做一次结构优化,看是否达到优化精度! 对策2:用这个CONTCAR拷贝成POSCAR,并且修改EDIFF(目前参数EDIFF=1E-6),默认为10-4 错误: WARNING:Sub-Space-Matrix is not hermitian in DAV1-7.626640664998020E-003 网上参考解决方案: 对策1:减小POTIM:IBRION=0,标准分子动力学模拟。通过POTIM控制步长。 POTIM:当IBRION=1,2或3时,是力的一个缩放常数(相当于确定原子每步移动的大小),默认值为0.5。 对策2:改IBRION=1,采用准牛顿算法来优化原子的位置。 原IBRION=2,采用共轭梯度算法来优化原子的位置 对策3:修改ISMEAR 对策4:换成CG弛豫(共轭梯度算法)IBRION=2(决定结构优化过程中,原子如何移动或弛豫) IBRION=2离子是否运动,1不运动但做NSW外循环。0动力学模拟,1准牛顿法离子弛豫 2CG法离子弛豫,3采用衰减二阶运动方程离子弛豫, INCARrelax中设置IBRION=2,未解决! 对策5:用的CG算符,出现的错误是CG算符不能算,在INCAR中加上IALG=Fast(电子优化采用blocked Davidson 方法[IALGO=38:IALG=Normal]和RMM-DIIS算法[IALGO=48:IALG=Very_Fast]混合)试一试
自旋-轨道作用[编辑] 在量子力学里,一个粒子因为自旋与轨道运动而产生的作用,称为自旋-轨道作用(英语:Spin–orbit interaction),自旋-轨道效应或自旋-轨道耦合。最著名的例子是电子能级的位移。电子移动经过原子核的电场时,会产生电磁作用.电子的自旋与这电磁作用的耦合,形成了自旋-轨道作用。谱线分裂实验明显地侦测到电子能级的位移,证实了自旋-轨道作用理论的正确性。另外一个类似的例子是原子核壳层模型(shell model)能级的位移。 半导体或其它新颖材料常常会涉及电子的自旋-轨道效应。自旋电子学专门研究与应用这方面的问题。 目录 [隐藏] 1 电子的自旋-轨道作用 1.1 磁场 1.2 磁矩 1.3 哈密顿量微扰项目 1.4 能级位移 2 参阅 3 参考文献 4 外部链接 电子的自旋-轨道作用[编辑] 在这篇文章里,会以相当简单与公式化的方式,详细地讲解一个束缚于原子内的电子的自旋-轨道作用理论。这会用到电磁学、非相对论性量子力学、一阶微扰理论。这自旋-轨道作用理论给出的答案,虽然与实验结果并不完全相同,但也相当的符合。更严峻的导引应该从狄拉克方程开始,也会求得相同的答案。若想得到更准确的答案,则必须用量子电动力学来计算微小的修正。这两种方法都在本条目范围之外。 磁场[编辑] 虽然在原子核的静止参考系 (rest frame) ,并没有磁场;在电子的静止参考系,有磁场存在。暂时忽略电子的静止参考系不是惯性参考系,则根据狭义相对论[1],磁场是 ;(1) 其中,是电子的速度,是电子运动经过的电场,是光速。 以质子的位置为原点,则从质子产生的电场是 ; 其中,是质子数量(原子序数),是单位电荷量,是真空电容率,是径向单位矢量,是径向距离,径向矢量是电子的位置。 电子的动量是 ; 其中,是电子的质量。 所以,作用于电子的磁场是 ;(2)
ADF教程:如何计算自旋-轨道耦合矩阵元SOCMEs 前言: 自旋-轨道耦合对于磷光很重要,因为如果二者耦合如果严格为0,那么单重态和三重态之间的跃迁就会成为禁阻跃迁,就不会有磷光发生。 有时候我们需要关心某个特定几何结构下(例如S0态与T1态势能面交叉点处),S0态与T1态之间自旋轨道耦合。用算符来表示即:
Bi2Se3自旋轨道耦合计算
Bi2Se3自旋轨道耦合性质的计算 一、模型和基本参数: 图(a)黑色t1、t2、t3基矢围成Bi2Se3菱形原胞,用于计算块体,红色方框包含一个五元层,是构成薄膜的一个QL。 计算能带的布里渊区高对称点:Г(0 0 0)-Z(π π π)-F(π π 0)-Г(0 0 0)-L(π 0 0), 根据正空间和倒空间坐标的转换关系, 得到正空间中高对称点的坐标:Г(0 0 0)-Z(0.5 0.5 0.5)-F(0.5 0.5 0)-Г(0 0 0)-L(0 0 -0.5) 空间群:166号~ R-3M(MS))3(5 3 m R D d (文献)
结构分为:六角晶胞和菱形原胞(Rhombohedral)两种形式 六角晶胞(hexagon):含三个五元层,15个原子菱形原胞(Rhombohedral):含5个原子 晶格参数t=9.841, α=24.275 原子坐标: 弛豫值实验值 Bi(2c) (0.400,0.400,0.400) Bi(2c) (0.398, 0.398, 0.398) Se(1a) (0,0,0) Se(1a) (0,0,0) Se(2c) (0.210, 0.210, 0.210) Se(2c) (0.216, 0.216, 0.216) 赝势:PAW_GGA_PBE E cut=340 eV 块体:Kpoints=11×11×11 薄膜:Kpoints=11×11×1 块体结构优化时,发现Ecut=580,KPOINTS=151515,得到的结构比较合理 计算薄膜真空层统一:15 ?
ISMER取-5(或取0,对应SIGMA=0.05)二、计算过程描述: 1)范德瓦尔斯作用力的影响。 手册中一共有5种方法: Correlation functionals:LUSE VDW = .TRUE. the PBE correlation correction AGGAC = 0.0000 Exchange交换functionals vdW-DF vdW-DF2 方法一方法二方法三方法四方法五revPBE optPBE optB88 optB86b rPW86 GGA = RE LUSE_VDW = .TRUE. AGGAC = 0.0000 GGA = OR LUSE_VDW = .TRUE. AGGAC = 0.0000 GGA = BO PARAM1 = 0.1833333333 PARAM2 = 0.2200000000 LUSE_VDW = .TRUE. AGGAC = 0.0000 GGA = MK PARAM1 = 0.1234 PARAM2 = 1.0000 LUSE_VDW = .TRUE. AGGAC = 0.0000 GGA = ML Zab_vdW = -1.8867 LUSE_VDW = .TRUE. AGGAC = 0.0000 经测试,发现方法二optimized Perdew-Burke-Ernzerhof-vdW (optPBE-vdW)是最合适的。 并通过比较发现,范德瓦尔斯作用力对块体和单个QL厚度的薄膜的影响很小,对多个QL厚度的薄膜结构影响比较大,所以优化时需要考虑QL之间的vdW相互作用,而范德瓦尔斯作用力对电子态的影响也比较小,所以,计算静态和能带的时候,可以不考虑。 此外,以往文献中的计算,有的直接采用实
如何计算自旋-轨道耦合矩阵 前言: 自旋-轨道耦合对于磷光很重要,因为如果二者耦合如果严格为0,那么单重态和三重态之间的跃迁就会成为禁阻跃迁,就不会有磷光发生。 有时候我们需要关心某个特定几何结构下(例如S0态与T1态势能面交叉点处),S0态与T1态之间自旋轨道耦合。用算符来表示即:
第二步,进行自旋-轨道耦合矩阵元的计算。这一步计算的物理意义:首先以Scalar相对论(无自旋轨道耦合的相对论方法)将较低的单重激发态和三重激发态计算出来,然后将自旋-轨道耦合视为微扰,得到自旋-轨道耦合矩阵元,然后也得到考虑微扰之后的各个激发态的激发能(此时,三重态可能会发生劈裂,即三个态能量不等——这就是由自旋-轨道耦合引起的)。 因此,计算参数设置如下: