
能带结构和态密度图的绘制及初步分析
前几天在QQ的群中和大家聊天的时候,发现大家对能带结构和态密度比较感兴趣,我做计算已经有一年半了,有一些经验,这里写出来供大家参考参考,希望能够对初学者有所帮助,另外写的这些内容也不可能全都正确,只希望通过表达出来和大家进行交流,共同提高。
MS这个软件的功能确实是比较强,但是也有一些地方不尽如人意的地方。(也可能是我对一些结果不会分析所致,有些暂时不能解决的问题在最后一部分提出,希望大家来研究
研究,看看有没有实现的可能性)。
能带结构、态密度和布居分析是很重要的内容,在
分析能带结构和态密度的时候,往往是先作图,然后分
析。
软件本身提供的作图功能并不是很强,比如说能带结构
(只能带只能做point图和line图),不美观不说,对于
每一个能带的走势也不好观察,感觉无从下手。所以我
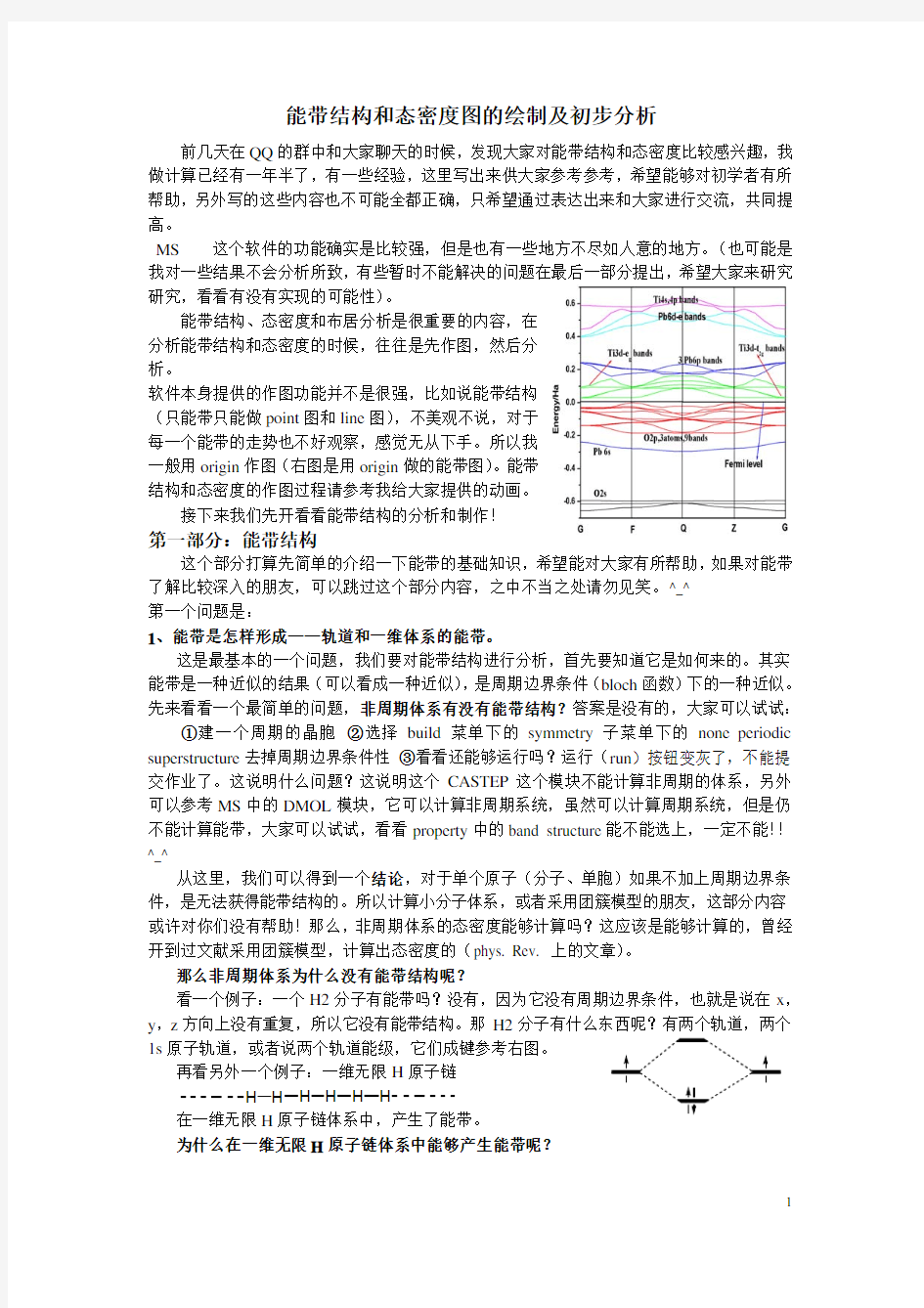
一般用origin作图(右图是用origin做的能带图)。能带
结构和态密度的作图过程请参考我给大家提供的动画。
接下来我们先开看看能带结构的分析和制作!
第一部分:能带结构
这个部分打算先简单的介绍一下能带的基础知识,希望能对大家有所帮助,如果对能带了解比较深入的朋友,可以跳过这个部分内容,之中不当之处请勿见笑。^_^
第一个问题是:
1、能带是怎样形成——轨道和一维体系的能带。
这是最基本的一个问题,我们要对能带结构进行分析,首先要知道它是如何来的。其实能带是一种近似的结果(可以看成一种近似),是周期边界条件(bloch函数)下的一种近似。先来看看一个最简单的问题,非周期体系有没有能带结构?答案是没有的,大家可以试试:
①建一个周期的晶胞②选择build菜单下的symmetry子菜单下的none periodic superstructure去掉周期边界条件性③看看还能够运行吗?运行(run)按钮变灰了,不能提交作业了。这说明什么问题?这说明这个CASTEP这个模块不能计算非周期的体系,另外可以参考MS中的DMOL模块,它可以计算非周期系统,虽然可以计算周期系统,但是仍不能计算能带,大家可以试试,看看property中的band structure能不能选上,一定不能!!^_^
从这里,我们可以得到一个结论,对于单个原子(分子、单胞)如果不加上周期边界条件,是无法获得能带结构的。所以计算小分子体系,或者采用团簇模型的朋友,这部分内容或许对你们没有帮助!那么,非周期体系的态密度能够计算吗?这应该是能够计算的,曾经开到过文献采用团簇模型,计算出态密度的(phys. Rev. 上的文章)。
那么非周期体系为什么没有能带结构呢?
看一个例子:一个H2分子有能带吗?没有,因为它没有周期边界条件,也就是说在x,y,z方向上没有重复,所以它没有能带结构。那H2分子有什么东西呢?有两个轨道,两个
1s原子轨道,或者说两个轨道能级,它们成键参考右图。
再看另外一个例子:一维无限H原子链
H H H H H H
在一维无限H原子链体系中,产生了能带。
为什么在一维无限H原子链体系中能够产生能带呢?
因为,每一个H 原子有一个1s 轨道,由于在X 轴方向(H 原子周期排列的方向)引入周期边界条件,所以这个体系有无数(阿佛加得罗)个H1s 的轨道能级,这些具有相同能量的能级轨道处于简并的状态。如果两个相邻的H 原子之间距离较大,不能够成键,那么这无数个简并的能级将排成一条水平的直线,这条直线很长,无法画下来,那么我们只有压缩它,将他压缩到一个区间([0,a π
]),这样每一个能级用一个点表示,由于点较多,看起
来好像形成了一条线,这样能带就形成了。
如果用函数的语言来描述,周期排列我们采用bloch 函数表示,我们解这些函数,就得到了一些k 矢量,在一维体系中k 矢量表示平移操作,k 的取值见下图,由于H1s 轨道能级有无数个,所以k 平移操作矢量就有无数个(注意k 是量
子化的),所以将它们压缩到[0,a π
]这个区间,就成了能带
结构中的横坐标,另外这个矢量也可以指向-X 方向,所以在[-
a π,0]这个区间能带的图像和[0,a π
]对称。
当H 原子之间的距离逐渐接近,它们的原子轨道要进行组合,形成一个成键分子轨道和一个反键分子轨道,那么原来能带是一条水平的直线,现在就要开始发生弯曲了(两个分子轨道能量不一样,导致能带发生弯曲),所以[0,a π
]这个区间,能带开始有带宽(散度),
随着H 原子的距离逐渐接近,可以预料,成键分子轨道和反键分子轨道的分裂越大,能带的带宽(最高能级-最低能级)越大。所以,相邻轨道之间的重叠越大,成键程度越大,带宽就越大。
另外值得一提的是,k 矢量还可以表示节点的数目。当k =0的时候表示什么呢?表示节点数为0,没有节点,所以k =0表示的H1s 轨道组合(组成分子轨道的原子轨道都带+号)具有最大的成键,能量最低。随着k 值的增大,节点数逐渐增多,体系的能量上升,最后k =a π
时,H1s 轨道组合成的分子轨道能量最高(原子轨道为+-+-……交替)。所以H 原子链的能带结构是一条向上弯曲的曲线(能带),k =0能量最低,k =a π
时能量最高。
这里要特别注意,并不是k =0的时候节点数都为0,比如(+-,+-,+-,……)这样的p 轨道,如果它们沿着X 轴周期排列,那么k =0的时候将具有最大的节点数,这时候形成的分子轨道将是能量最大的,随着k 的逐渐增加,节点数逐渐减小,所以这时候能带将向下伸展,这与H 原子链的情况刚好相反。
大家都知道,这样的H 原子链是不可能稳定的,最后都要变成H2分子,能带要消失,这是一个什么样的过程呢?在这个一维周期体系中轨道能级的数目假设为N (无限大),那么这个体系的电子数是多少呢?答案是N ,那么这些电子在这个能带上是如何分布的呢?当然是按照能量从低到高的顺序来填充的了,这样由于每一个轨道能够容纳2个电子,而这个能带只有较低能量的部分被填充(能带半充满),所以这时候要产生畸变(Peierls 畸变,即固体物理中的姜-泰勒效应),H 原子之间要产生相对振动(虚模,能量不稳定),以降低体系的能量,这样,H2分子就形成了,而能带也由于H2原子的形成破坏了周期条件,当这些H2分子不再沿着X 轴方向形成周期排列的时候,能带也就消失了(变成非周期体系)。
结论:一个原子的一个原子轨道在一维周期条件下将产生一条能带,能带的带宽取决于这些原子轨道的在周期方向上的成键强度,强度越大,带宽越大,成键越弱,带宽越小,如
果周期方向上没有成键,能带将是一条直线。另外能带是向上伸展还是向下伸展取决于原子轨道的特性,或者说是体系的拓补性质。
接下来我们看看,布里渊区里面的高对称点(G ,X ,F ,M 等)是怎么来的。
2、 布里渊区的高对称点
前面讲了一维周期条件下的布里渊区的能带是一条线,如果加上二维(X ,Y )的周期边界条件,这些能带又会变成什么样呢?答案是一个面,由原来的线组成一个面。因为在一维情况下我们用一条能带来表示k 矢量(对称操作)和能级的关系,可以用E (k )来表示,这构成第一布里渊区(即k 的取值范围[0,a π
])。对于二维周期体系,我们需要两个平移矢
量k x 和k y ,所以能带可以用E (k x ,k y )来表示,当k x =0时,变成E (0,k y ),得到一条能带(线,y 方向上与一维周期情况的能带类似);当k y =0时,变成E (k x ,0),得到一条能带(线)。由于k x 和k y 是矢量,它们可以组合成另外一个矢
量,这个矢量不是沿着X 轴,也不是沿着Y 轴,实际上沿着该
矢量仍是能够得到一个能带(线)的,这样的矢量有很多,所
有的这些能带(线)将构成一个面。如果我们在做能带结构图
的时候,将能带结构按照二维的面画出来是很困难的,而三维
的情况更加困难,因为对称操作有很多,k 矢量的取值有很多,
所以一个可行的办法就是让k 的取值沿着一定的路径走,最后
回到起点。如右图(二维情况)。
这样,我们只要选择一些较高的对称点,就可以确定这个路径。比如二维的布里渊区是一个面,这个面上每一点与原点(G 点或Γ点)的连线都构成一个k 矢量,有一个k 矢量就有一个能级对应(E (k ))。所以,二维的能带结构是这个布里渊区上的一个平面(面积),如上图,按照Γ——>X ——>M ——>Γ这个路径走,就可以得到一个可以大致反映布里渊区上的能带平面的一个近似图,这就是二维的能带结构。具体的能带图的展开见下图。
三维的能带展开见下图:
前面讲了一维H 原子链的能带结构,提到这个体系具有一个能带,并且是向上弯曲的一个能带。这个能带是这样来的,H 原子链里面的基本单位(单体,单胞)是一个H 原子,每一个原子有一个原子轨道,即每一个基本单位有一个能级轨道,加上周期条件以后,这一系列轨道能就变为一个能带了。假如,现在以两个H 原子作为一个基本单位呢?能带结构又是如何的呢?这就是能带重叠的问题
3、 能带的折叠
如上图,将2个氢原子作为一个基本单位,这时候能带结构是什么样子的呢?很容易理解,根据前面的知识,单胞的原子轨道的数目决定了能带的数目,所以这样划分体系将有将有两个能带。
但是这个体系与前面1个H 原子周期链是一样的,只不过人为地进行了划分,能带结构就变了吗?是的,能带确实变了,那么能带将怎样变化呢?下图分别是1个H 原子为单胞和2个H 原子为单胞的能带结构。
第一个图可以清楚的看到,能带底部是成键的,能带的顶部是反键的,中间是非键的(成键与反键相当),能带向上伸展(弯曲)。在第二个图,原来的中部的非键轨道分别变成了两个能带的反键和成键轨道(相同颜色表示可以重叠成键,不同颜色表示中间有一个节点)。实际上,这两个图是有关系的,能够反映相同的内容,首先,布里渊区从原来的[0,
a π]变为[0,a 2π
],这也是可以理解的,原来的能带长度要变成原来的一半(因为周期方向上的单胞数减少一半,原来有N 个单胞,以2个H 原子为一个单胞后,单胞数变为一半,所以布里
渊区要减半)。其次,原来的能带在[0,a π
]展开,现在由于布里渊区减半,能带不能在[0,a 2π
]
这个小区间画出来,所以能带结构将产生折叠,由原来的一个能带变为2个能带,并且是以k =a 2π
为对称轴,将原来[a 2π,a π
]区间的能带折叠过去,所以就得到了2H 原子为单胞的
能带结构。如果我们将单胞取3个原子,或者取4个原子,体系的能带将如何变化?体系的能带分别变为3条和4条能带,并且是2次或3次折叠原来的能带。
另外,上面的例子我们也可以从另外一个角度考虑问题,即2个H 原子组成一个H2分子,形成成键分子轨道和反键分子轨道,把这两个分子轨道看成一个“原子”的两个“原子轨道”,给这个“原子”加上周期条件以形成能带,这
样也得到2个能带。这种想法可以进行适当的扩充,
比如我们研究一个晶体,首先我们可以把单胞找出来,
然后将单胞的轨道能级画出来,考虑它们之间的成键,
最后得到一组分子轨道,以这组分子轨道为基础,给
它们加上周期条件,形成能带。这样的想法什么时候
有用呢?在组合成分子轨道以后,这些分子轨道并不是
不变的,有可能和相邻晶胞的分子轨道再次组合,这
种情况在过渡金属氧化物中非常普遍,所以在考虑成
键的时候除了单胞内成键情况以外,还需要考虑晶胞间分子轨道的组合。
能带的折叠实际上是从不同的角度考虑问题而已,其实能带结构的本质还是一样的。如果我们理解了这点,那么我们在分析能带的时候,就不会笨到建立一个很大的超胞去研究其能带结构。所以我们要研究一个体系的能带结构时候,尽量选最小的单胞,以减少能带的数目。
不幸的是,有时候我们往往要计算掺杂的情况而不得不建立一个很大的单胞(超胞),而掺杂的比例又与超胞的大小有关。如果要做1/8掺杂,那么起码要将8个晶胞做成一个单胞(超胞),并将其中一个原子替换掉,从而实现掺杂。这样能带的数目将变为原来的8倍,这样我们计算出来的能带将是一团曲线交缠在一起,再也无法分开。
大家可以试试建立一个超胞,然后计算能带,看看能带结构如何!
所以能带结构的分析对于比较大的单胞(超胞)或者具有较多原子轨道的单胞将变得很麻烦。而且MS 中画出的能带结构很难看,如果我们要用origin 作图的画,又要反复的copy +paste 数据,所以建议在计算具有较多原子轨道(分子轨道)的体系时,尽量不用能带结构分析问题(虽然它很有用^_^)。用态密度和PDOS 就好多了。
另外,简单的提一下Fermi 能级的概念,有很多书都将最高占据分子轨道(HOMO )的顶部作为Fermi 能级,所以我也采用这种观点,不知道大家是怎么理解的?
下面我简单讲讲我做的体系的能带结构,具体做法参加我提供的动画演示。
4、3维体系的能带——PbTiO3的能带结构
要分析能带,首先要知道每个能带是属于那个轨道的,所以首先要画出单胞的轨道能级图,下图是钛酸铅的轨道能级示意图,不是很准确(因为有一些轨道成键没有连线,仅给价带和导带部分连线了)。
在轨道能级图里面先将各个原子的轨道能级标出来(横轴的能量是参考了PDOS 加上的,没有也没有关系^_^)。接着考虑成键,主要是中心Ti 原子和O 原子成键(模型参考下图),有O 原子的2p 轨道组成价带,2s 轨道的能量最低Pb 的4s 轨道比O2p 轨道能量低,比O2s 轨道高。另外Ti 原子处于TiO6八面体中心,所以3d 能级要分裂成3下(3t 2g 态,3d xy ,3d xz ,3d
yz 单胞1单胞2
二次组合
轨道)和2上(e g 态,即3d x 2-y 2,3d z 2轨道),这些轨道构成导带。Pb 的6d ,Ti 的4s ,4p 轨道在Ti3d 轨道上面。
有了这些信息我们就可以绘制能带,并可以用不同的颜色表示不同轨道形成的能带,具体的绘制过程见动画。
接下来看看能带能够说明什么问题。
第一点:O2s 能带有带宽,表明其参与成键(参与成
键的有Ti 的3d z 2轨道,Ti4s ,4p z 轨道,还有Pb 原子轨道
的部分贡献,能带结构看不出来,但是态密度可以看出
来)。
第二点:Pb 的6s 能带带宽比较大,说明Pb6s 轨道参
与成键,由于Pb 与Ti 原子很远(3.45A )所以只能是与
O2p 轨道成键(能量相近,并且有带宽,从后面态密度分
析也可以得到同样的结论)
第三点:O2p 能带组成价带,体系中有3个O 原子,
共有9条O2p 能带,其中每3个是一组(G 点是3重简并
的)。
第四点:Ti3d 的5个能级分裂成2组,3下2上,组成导带。
第五点:Pb 的6p 和6d 能带在Ti 的3的能带上面,而且Pb 的6的也要分裂,还有一些能带图中未能表示出来,另外Ti4s ,4p 能带在最上面,且带宽比较大,说明它们参与了与O2p 轨道成键。
当然,能带结构还能给出很多信息,比如带隙等,由于水平有限,所以只能看出一些简单的内容!希望对能带结构比较了解的朋友能够把这部分内容更加完善,大家共同探讨和学习!
下面我主要讲一下态密度的一些基本内容和初步的分析以及态密度的绘制。 第二部分:态密度
首先来看一下态密度的意义,前面提到当单胞(超胞)很大的时候,含有的原子轨道(分子轨道)的数目就很多,采用能带分析的时候,必须处理很多能带。在单个分子中,我们可以挑选出一个或一小组能决定分子的几何构型,反应性能等的轨道作为前线轨道或者价轨道,但是,在多到几乎无法可数的晶体轨道(组成能带的能级)中,就没有可能找到某一个能级来决定一种几何构型或反应性能。
举个例子来说,如果做表面成键或者吸附,首先要建立按照一定的晶面(如[110]面)取向的表面构型,然后就是考虑吸附上去的原子对这个表面的影响情况,如果计算能带,当
表面吸附的覆盖度很小时,这时候需要建的表面就很大,能带的数目自然就增加很大数目,而影响到表面结构和性质的只要那么几条能带(吸附原子和被吸附原子的能带),如何从这么多的能带中找到它们呢,这显然时吃力不讨好的事情(我觉得这时候用能带来解释不现实),那么我们怎么考虑吸附质吸附上去以后,表面的结构和性质呢。我个人认为,只能靠计算得到的态密度来分析了,特别时偏态密度(态密度投影或称PDOS),比较被吸附原子和吸附原子在吸附前或者吸附后的偏态密度应该能够很好的说明情况,而且如果能够加上布居分析,局部的结构和性质应该可以弄清楚。比如做TiO2[110]面吸附乙酸,我们可以计算TiO2晶体的态密度,计算TiO2[110],和吸附后的TiO2[110]的态密度,这样,比较前两者,就知道从晶体变到表面以后,吸附原子的成键变化情况,比较后两者,又可以知道吸附前和吸附后,表面的结构怎么变化(成键等),性质发生什么变化。所以我个人认为,在计算表面的结构性质的时候,应该更注意局部变化的情况。
那么态密度的物理意义是什么呢?
1、态密度的意义
态密度的物理意义应该是比较简单的。大家都知道,能带结构的纵坐标是能量,假如在这个坐标轴上取ΔE这一个很小能量范围(比如0.005000~0.005001eV这个范围),那么这个能量间隔范围内又多少个能级,或者说又多少个原子轨道(分子轨道)呢?别忘了,能带是怎么来的,是无数个能级“压缩”而成的,而且能带是量子化的,所以在这个能量范围必然又一定数量的能级(轨道)存在。所以从这里开始,不再谈论单个的原子轨道或能级了,因为这没有什么意义。而是我们将这一能量范围内的能级作为一组来考虑,所以态密度的概念就得出来了,即E+dE这个能量范围内的能级数,如果E+dE这个能量范围内轨道(能级数)越多越密集,态的密度越大,所以说态密度的概念是很贴切的。
而且要注意态密度是根据能带得来的,两者有一定的对应关系,比如在E+dE这个能量间隔没有能带,那么有态密度吗?这个答案是很显然的,没有能带,没有能级,哪能有态密度呢?所以这时候态密度图在这个能量范围内将是0。从这里可以得到一个结论,能带按照纵坐标轴投影过去,就得到态密度,所以态密度为0的地方,能带图上一定没有能带经过。
在态密度中还有一个问题就是,有的峰很高,有的峰很低,这是为什么呢?为什么会产生这种情况?这个很容易理解,比如一条水平的能带经过E+dE这个区间,那么这个区间的能级数是不是最大的呢?当然是,这个能带的所有能级都处于这个能量范围内,所以态的数目最大,在态密度图中表现为一个很尖锐的峰。如果能带的带宽较大,这说明态密度图中它很平缓,并且跨过的能量区间越大。一句话,能带越平,态密度峰越尖锐,能带越宽,态密度越平缓,当然离域性越强了。
这里有两个问题:第一个问题:能带越平,是否成键越弱,或者不成键?即态密度峰越尖锐,成键越弱或不成键呢?第二个问题:能带带宽越大,即态密度峰跨度越大越平缓,成键是否越强?
这两个问题,我查了一些书,但是没有答案,我个人的理解如下。对于第一个问题,能带越平,不表示不成键或成键弱,为什么呢,比如两个原子轨道形成很强的σ键,那么根据分子轨道理论,将形成一个成键分子轨道和一个反键分子轨道,成键越强,两者分离越大,形成这两个分子轨道以后,在这个基础上在形成能带结构,也就是在X,Y,Z方向上加上周期条件,假如在这3个周期方向单胞之间的距离比较远,那么两个分子轨道不能产生2次组合,所以得到的能带只能是两条水平直线,所以我个人认为,能带平缓,或者态密度峰尖锐不能说明这个轨道不成键或者成键比较弱。
第二个问题,我认为是这样的,带宽越大,成键越强,因为带宽越大,态密度峰跨度越大,离域性越强,虽然不一定跟那个原子成键,但是其上面的电子离域了,肯定要成键,离域越强,成键越强。
上面两个问题是我自己思考的答案,当然不一定对,希望大家能够探讨探讨!
接下来,想说说用态密度如何分析成键的问题。
2、 态密度与成键
下面的图是能级,能带以及态密度的关系,两个原子的原子轨道组合以后,得到两个分子轨道,在周期边界条件下,这两个分子轨道形成两个能带,根据能带的宽度和斜率,可以得到态密度的近似图。
上图右边的态密度图表示的是总的态密度,不是PDOS ,总态密度与PDOS 的关系如何呢?要解决这个问题,还得回到能带结构,在能带结构中,同一个能量E 画一条水平横线,与能带相交,交点有可以能很多个,并且有可能和几条能带相交,与这条直线相交的若干个点对应于态密度图上能量为E 的一个点,也就是说这个点包含了若干个能带的贡献,所以得到总的态密度图,如果非要区分这些交点中属于那些轨道,则总态密度就得按照一定的原则子划分到不同原子上,就得到了PDOS 。总之,总态密度是所有能带的贡献,而如果要对这些贡献划分为某一个原子的贡献,则产生PDOS 。
再回到上面的图,上图中左边为分子轨道,这个分子轨道可以用波函数2211χχψc c +=来描述,其中1χ和2χ表示两个原子轨道,那么它们组成的每一个分子轨道应该都有两个原子轨道的贡献。但是对于成键轨道,主要以电负性大的原子的原子轨道为主(比如O2p 轨道),即成键分子轨道主要是电负性大的原子的原子轨道,当中掺入了部分另外一个原子轨道的贡献;而反键分子轨道主要是由电负性小的原子的原子轨道构成,并且有电负性大的原子轨道的贡献。也就是所这两个能带不是某一个原子轨道独自形成的的,每一个能带都是一个原子轨道为基础,掺入了其他轨道的贡献,当然这个性质在能带结构中是无法反映的,你不知道其他原子的原子轨道对某一个能带的贡献。那么,什么能够反映这个性质呢?答案是态密度,更准确一些讲是PDOS 。下面看看如何用PDOS 表示这个性质!
如下图:
这个图有两部分态密度组成,能量较低的部分态密度相当于低能的能带产生,高能部分的态密度是高能能带产生(参考上面的轨道-能带-态密度图)。这样低能部分的态密度对应于成键分子轨道,它由两个部分组成(红色部分+黑色部分=总态密度),其中面积大的部分态密度是由电负性大的原子的原子轨道产生,而面积小的部分是另外一个原子轨道对这个分子轨道的贡献(对低能能带的贡献),而对于高能部分的态密度刚好相反,所以根据PDOS可以判断成键的情况。也就是说态密度发生“共振”是成键的一个明显标志。
对于态密度,另外还有一些东西要注意的:
第一:对态密度曲线的积分等于电子数,比如体系由10个电子,10个电子肯定是按照能量从低到高的顺序排列,那么对态密度进行积分,当电子数达到10的时候,这个地方就是fermi能级(积分曲线可以用origin做)。
第二:偏态密度积分至fermi能级得到某一个原子某一个轨道的电子填充的数目。
第三:如果成键作用加强,那么成键分子轨道要下移,反键分子轨道要上移,导致态密度要发生移动,一个向下移动,一个向上移动,而能带则变宽。
大家如果要验证态密度的移动,可以计算一些亚晶格的态密度,然后组合,看看移动的方向。比如钛酸铅,去掉Ti原子和Pb原子,得到O3的亚晶格,计算它的态密度;去掉O3和Ti计算Pb原子的亚晶格,最后用同样方法计算Ti原子的亚晶格的态密度。根据算出的结果和PbTiO3的态密度比较,会发现很明显的特征,起码态密度的移动很明显,而且轨道要分裂(如Ti分裂成3下2上结构),等等!
从上面的讨论和分析可以清楚的知道,PDOS具有很大的用处,不但可以分析成键,也可以分析电子在何处(对不同PDOS积分)!
下面看看我计算的钛酸铅的态密度:
上面的左图是Ti-O原子之间的成键分析,可以发现O2p轨道和Ti3d轨道有明显的“共振”,另外Ti4p在O2p有一部分贡献;Ti4s,4p,3d对O2s都有态密度贡献(考虑到对称性匹配,它们应该成σ键)
上面的右图可以发现,Pb的6s和O2p有态密度共振,也成键;另外Pb6d和Pb6d在O2p态密度处有明显的峰(有贡献),所以O2p与Pb6s,6d也是成键的。
虽然,态密度能够分析成键的情况,并且能够积分出电子的数目从而确定电子在原子轨道上的分布情况。但是对于成键的强弱很难以定量说明,也就是说,成键强,到底有多强,成键弱,到底有多弱这样的问题用态密度比较难以回答。一个办法就是积分PDOS的贡献峰,但是,这样做也不见得可行,比如大家看看上面O2p的贡献峰,O2p在Pb6s的贡献峰积分,就可以知道它与Pb6s轨道成键时对Pb6s轨道的贡献。但是由于Ti3d和Pb6p态密度
峰重叠了,O2p对这两个轨道的贡献峰是一个总的贡献,区分不开对Ti3d和Pb6p的贡献。
态密度对成键的定量化描述不是很强,所以我们往往要进行布居分析,原子布居和键布居,根据布居的数值来判定成键的强弱。
态密度的基本内容就这些,至于怎么作图,可以用origin做,大家看看我提供的动画,动画中有部分内容是积分曲线的制作。
第三:MS中Castep和Dmol模块的一些问题
最后,想讲使用MS中的一些问题提出来,由于我一直没有解决,在这里提出,大家看看能不能实现。
第一个问题就是MS中能不能做晶体轨道重叠布居图(COOP图)?
前面提到用DOS和PDOS 分析成键,对于其强弱的变化比较难以描述,所以要借助布居分析,而晶体轨道重叠布居图,就是重叠布居权重的态密度。我们还是那1维H原子链来说明问题,如下图:
在这个体系中,能带下面是成键的,中间是非键的,上部是反键的。当电子填充在能带下部的时候,填充越多成键越强,当下半部分能带电子填满以后,在往上填充,电子就会跑到反键轨道上,使得成键逐渐减弱。COOP图能够很直观的反映出成键的情况,是一个分析成键情况的有力工具,可是MS中好像做不出来,但是ADF有这个功能,所以在这个地方提一下,大家看看MS中有没有实现的可能性。
第二个问题就是重叠布居的问题。重叠布居是键强弱的一个度量,重叠布居本身是有很多轨道贡献的叠加,这里提出这个问题是MS中能够把没有原子轨道对重叠布居的贡献烈出来吗?
另外还有一些问题,以后慢慢在论坛或者群里面在问好了。^_^
另外,强烈推荐一本书,上面的一些观点都是从这本书中找到更为详细的解释。
书名:固体与表面
作者:[美国] R.霍夫曼著
郭洪煪李静译
王作新郑冲校
出版社:北京工业出版社
我的联系方式:carlon@https://www.doczj.com/doc/c92914311.html,或carlon2000@https://www.doczj.com/doc/c92914311.html,
R语言与非参数统计(核密度估计) 背景 核密度估计是在概率论中用来估计未知的密度函数,属于非参数检验方法之一,由Rosenblatt (1955)和Emanuel Parzen(1962)提出,又名Parzen窗(Parzen window)。 原理 假设我们有n个数X1-Xn,我们要计算某一个数X的概率密度有多大。核密度估计的方法是这样的: 其中K为核密度函数,h为设定的窗宽。 核密度估计的原理其实是很简单的。在我们对某一事物的概率分布的情况下。如果某一个数在观察中出现了,我们可以认为这个数的概率密度很大,和这个数比较近的数的概率密度也会比较大,而那些离这个数远的数的概率密度会比较小。基于这种想法,针对观察中的第一个数,我们都可以f(x-xi)去拟合我们想象中的那个远小近大概率密度。当然其实也可以用其他对称的函数。针对每一个观察中出现的数拟合出多个概率密度分布函数之后,取平均。如果某些数是比较重要,某些数反之,则可以取加权平均。 但是核密度的估计并不是,也不能够找到真正的分布函数。 代码作图示例 我们可以举一个极端的例子:在R中输入: ●[plain]view plaincopyprint? 1.plot(density(rep(0, 1000))) 可以看到它得到了正态分布的曲线,但实际上呢?从数据上判断,它更有可能是一个退化的单点分布。 但是这并不意味着核密度估计是不可取的,至少他可以解决许多模拟中存在的异方差问题。比如说我们要估计一下下面的一组数据: ●[plain]view plaincopyprint? 1.set.seed(10) 2.dat<-c(rgamma(300,shape=2,scale=2),rgamma(100,shape=10,scale=2))
高中数学知识点1 集合 123412n x A x B A B A B A n A ∈??? ????? ∈?∈?()元素与集合的关系:属于()和不属于()()集合中元素的特性:确定性、互异性、无序性集合与元素()集合的分类:按集合中元素的个数多少分为:有限集、无限集、空集()集合的表示方法:列举法、描述法(自然语言描述、特征性质描述)、图示法、区间法子集:若 ,则,即是的子集。、若集合中有个元素,则集合的子集有个, 注关系集合集合与集合{}00(2-1)23,,,,.4/n A A A B C A B B C A C A B A B x B x A A B A B A B A B A B x x A x B A A A A A B B A A B ?????????? ????????????≠∈?????=???=∈∈?=??=??=???真子集有个。、任何一个集合是它本身的子集,即 、对于集合如果,且那么、空集是任何集合的(真)子集。 真子集:若且(即至少存在但),则是的真子集。集合相等:且 定义:且交集性质:,,,运算{}{},/()()()-()/()()()()()()U U U U U U U U A A B B A B A B A A B x x A x B A A A A A A B B A A B A A B B A B A B B Card A B Card A Card B Card A B C A x x U x A A C A A C A A U C C A A C A B C A C B ????????=????=∈∈???=??=?=????????=???=+?=∈?=?=??==?=?,定义:或并集性质:,,,,, 定义:且补集性质:,,,, ()()()U U U C A B C A C B ????? ?? ?? ?? ?? ?????????? ???????? ??????????????????????? ?????????????????????=???????
能带图的横坐标是在模型对称性基础上取的K点。为什么要取K点呢?因为晶体的周期性使得薛定谔方程的解也具有了周期性。按照对称性取K点,可以保证以最小的计算量获得最全的能量特征解。能带图横坐标是K点,其实就是倒格空间中的几何点。其中最重要也最简单的就是gamma那个点,因为这个点在任何几何结构中都具有对称性,所以在castep里,有个最简单的K点选择,就是那个gamma选项。纵坐标是能量。那么能带图应该就是表示了研究体系中,各个具有对称性位置的点的能量。我们所得到的体系总能量,应该就是整个体系各个点能量的加和。 记得氢原子的能量线吧?能带图中的能量带就像是氢原子中的每条能量线都拉宽为一个带。通过能带图,能把价带和导带看出来。在castep里,分析能带结构的时候给定scissors这个选项某个值,就可以加大价带和导带之间的空隙,把绝缘体的价带和导带清楚地区分出来。 DOS叫态密度,也就是体系各个状态的密度,各个能量状态的密度。从DOS图也可以清晰地看出带隙、价带、导带的位置。要理解DOS,需要将能带图和DOS结合起来。分析的时候,如果选择了full,就会把体系的总态密度显示出来,如果选择了PDOS,就可以分别把体系的s、p、d、f状态的态密度分别显示出来。还有一点要注意的是,如果在分析的时候你选择了单个原子,那么显示出来的就是这个原子的态密度。否则显示的就是整个体系原子的态密度。要把周期性结构能量由于微扰裂分成各个能带这个概念印在脑袋里。 最后还有一点,这里所有的能带图和DOS的讨论都是针对体系中的所有电子展开的。研究的是体系中所有电子的能量状态。根据量子力学假设,由于原子核的质量远远大于电子,因此奥本海默假设原子核是静止不动的,电子围绕原子核以某一概率在某个时刻出现。我们经常提到的总能量,就是体系电子的总能量。
这个图是在MS中做的吗,如果是在选择分析能带结构时在对话框中做好将,点选成线,这样看能带会比较方便。 由于所计算的物质原胞分子数比较多,所以能带图会比较密集,然而我们在研究能带结构时,最关心的是费米能级附近的能带情况,对于其他的情况没有解读的必要。 当禁带宽度大于3ev时此物质一般为绝缘体,介于1 ,3之间时为半导体,小于1或者没有禁带宽度时为导体。能带的分析要与态密度结合进行分析,这样会知道哪些能带是有哪些能级或那些原子贡献的。 能带图的横坐标是在模型对称性基础上取的K点。为什么要取K点呢?因为晶体的周期性使得薛定谔方程的解也具有了周期性。按照对称性取K点,可以保证以最小的计算量获得最全的能量特征解。能带图横坐标是K点,其实就是倒格空间中的几何点。其中最重要也最简单的就是gamma那个点,因为这个点在任何几何结构中都具有对称性,所以在castep 里,有个最简单的K点选择,就是那个gamma选项。纵坐标是能量。那么能带图应该就是表示了研究体系中,各个具有对称性位置的点的能量。我们所得到的体系总能量,应该就是整个体系各个点能量的加和。 记得氢原子的能量线吧?能带图中的能量带就像是氢原子中的每条能量线都拉宽为一个带。通过能带图,能把价带和导带看出来。在castep里,分析能带结构的时候给定scissors这个选项某个值,就可以加大价带和导带之间的空隙,把绝缘体的价带和导带清楚地区分出来。 DOS叫态密度,也就是体系各个状态的密度,各个能量状态的密度。从DOS图也可以清晰地看出带隙、价带、导带的位置。要理解DOS,需要将能带图和DOS结合起来。分析的时候,如果选择了full,就会把体系的总态密度显示出来,如果选择了PDOS,就可以分别把体系的s、p、d、f状态的态密度分别显示出来。还有一点要注意的是,如果在分析的时候
第1章 空间几何体1 1 .1柱、锥、台、球的结构特征 1. 2空间几何体的三视图和直观图 11 三视图: 正视图:从前往后 侧视图:从左往右 俯视图:从上往下 22 画三视图的原则: 长对齐、高对齐、宽相等 33直观图:斜二测画法 44斜二测画法的步骤: (1).平行于坐标轴的线依然平行于坐标轴; (2).平行于y 轴的线长度变半,平行于x ,z 轴的线长度不变; (3).画法要写好。 5 用斜二测画法画出长方体的步骤:(1)画轴(2)画底面(3)画侧棱(4)成图 1.3 空间几何体的表面积与体积 (一 )空间几何体的表面积 1棱柱、棱锥的表面积: 各个面面积之和 2 圆柱的表面积 3 圆锥的表面积2 r rl S ππ+= 4 圆台的表面积22R Rl r rl S ππππ+++= 5 球的表面积2 4R S π= (二)空间几何体的体积 1柱体的体积 h S V ?=底 2锥体的体积 h S V ?=底31 3台体的体积 h S S S S V ?++=)31 下下上上( 4球体的体积 33 4 R V π= 第二章 直线与平面的位置关系 2.1空间点、直线、平面之间的位置关系 222r rl S ππ+=
2.1.1 1 平面含义:平面是无限延展的 2 平面的画法及表示 (1)平面的画法:水平放置的平面通常画成一个平行四边形, 锐角画成450,且横边画成邻边的2倍长(如图) (2)平面通常用希腊字母α、β、γ等表示,如平面α、平面β等,也可以用表示平面的平行四边形的四个顶点或者相对的两个顶点的大写字母来表示,如平面AC 、平面ABCD 等。 3 三个公理: (1)公理1:如果一条直线上的两点在一个平面内,那么这条直线在此平面内 符号表示为 A ∈L B ∈L => L α A ∈α B ∈α 公理1作用:判断直线是否在平面内 (2)公理2:过不在一条直线上的三点,有且只有一个平面。 符号表示为:A 、B 、C 三点不共线 => 有且只有一个平面α, 使A ∈α、B ∈α、C ∈α。 公理2 作用:确定一个平面的依据。 (3)公理3:如果两个不重合的平面有一个公共点,那么它们有且只有一条过该点的公共直线。 符号表示为:P ∈α∩β =>α∩β=L ,且P ∈L 公理3作用:判定两个平面是否相交的依据 2.1.2 空间中直线与直线之间的位置关系 1 空间的两条直线有如下三种关系: 相交直线:同一平面内,有且只有一个公共点; 平行直线:同一平面内,没有公共点; 异面直线: 不同在任何一个平面内,没有公共点。 2 公理4:平行于同一条直线的两条直线互相平行。 符号表示为:设a 、b 、c 是三条直线 a ∥ b c ∥b 强调:公理4实质上是说平行具有传递性,在平面、空间这个性质都适用。 公理4作用:判断空间两条直线平行的依据。 3 等角定理:空间中如果两个角的两边分别对应平行,那么这两个角相等或互补 4 注意点: ① a'与b'所成的角的大小只由a 、b 的相互位置来确定,与O 的选择无关,为了简便,点O 一般取在两直线中的一条上; ② 两条异面直线所成的角θ∈(0, ); ③ 当两条异面直线所成的角是直角时,我们就说这两条异面直线互相垂直,记作a ⊥b ; ④ 两条直线互相垂直,有共面垂直与异面垂直两种情形; ⑤ 计算中,通常把两条异面直线所成的角转化为两条相交直线所成的角。 2.1.3 — 2.1.4 空间中直线与平面、平面与平面之间的位置关系 1、直线与平面有三种位置关系: D C B A α L A · α C · B · A · α P · α L β 共面直线 =>a ∥c 2
密度图像和实验 1.甲、乙两种物质,它们的质量跟体积关系如图所示,则ρ _ _ρ乙(选填“>”、“<”或“=”),其中乙物质可能是____。 2.由同种某物质构成的大小不同的实心物块的体积、质量如下 表,以体积V为横坐标,以质量m为纵坐标,在图22的方格纸 一上描点,再把这些点连起来.该物质的密度是g/cm3. 3 、某物质的质量和体积的关系图象如图所示,体积是 20cm3的这种物质质量是多少?它的密度是多大? 4.给你一张密度表和一个可沉于水中的小铁球,请你再 自选其他器材,设计一个实验来判断该小铁球是实心 的还是空心的?简述你的实验方案. (1)主要器材:; (2)简要做法:; (3)如何判断:. 5.每当看到电视中出现演员被倒塌的房屋砸中,小明都 为演员捏一把汗。 现有以下器材:道具砖样品、一个量筒、一块体积为V铁的小铁块、一根细针、一段细线和足够的水。已知:道具砖不吸水,质地较软且密度小于水;水的密度为ρ水。为了测出道具砖的密度:(1)应从以上器材中选择的器材是:。 (2)实验步骤应为: (3)请用所测出的物理量写出道具砖密度的表达式,ρ= 。 6.下面是小丽在测量一块矿石密度时的主要步骤。 (1)下面的操作有哪些是必要的,请你把必要的操作按正确的顺序将序号排列出来:A.用调节好的天平测出量筒的质量为200g B.向量筒中倒进一些水,测出这些水的体积15 cm3 C.用调节好的天平测出矿石的质量28g D.将矿石浸没在量筒中,测出矿石和水的总体积25 cm3 E . 将必要的数据填入下表中,利用相应公式,求出矿石的密度。正确的操作顺序为。 2 7.在“用天平和量筒测量矿泉水密度”实验中,小明的实验步骤如下: (1)调好天平,测出空烧杯质量m1(2)在量简中倒入适量矿泉水,读出矿泉水的体积V (3)将量筒中矿泉水全部倒入烧杯中,测出矿泉水和烧杯总质量m2 则矿泉水密度的表达式ρ矿泉水= 以上操作由于无法将矿泉水从量简中倒尽,测出的矿泉水密度误差较大.经过思考,小明在
态密度(Density of States,简称DOS) 在DOS结果图里可以查瞧就就是导体还就就是绝缘体还就就是半导体,请问怎么瞧。理论就就是什么?或者哪位老师可以告诉我这方面得知识可以通过学习什么获得。不胜感激。 查瞧就就是导体还就就是绝缘体还就就是半导体,最好还就就是用能带图DOS得话瞧费米能级两侧得能量差 谢希德。复旦版得《固体能带论》一书中有,请参阅!另外到网上或者学校得数据库找找“第一性原理”方面得论文,里面通常会有一些计算分析。下面有一篇可以下载得:ZnO得第一性原理计算 hoffman得《固体与表面》对态密度得理解还就就是很有好处得。 下面这个就就是在版里找得,多瞧瞧吧: 如何分析第一原理得计算结果 用第一原理计算软件开展得工作,分析结果主要就就是从以下三个方面进行定性/定量得讨论:1 ?、电荷密度图(charge density); 2、能带结构(EnergyBand Structure);?3、态密度(Density ofStates,简称DOS)。??电荷密度图就就是以图得形式出现在文章中,非常直观,因此对于一般得入门级研究人员来讲不会有任何得疑问。唯一需要注意得就就就是这种分析得种种衍生形式,比如差分电荷密图(def-ormationchargedensity)与二次差分图(difference chargedensity)等等,加自旋极化得工作还可能有自旋极化电荷密度图(spin-polarizedc harge density)。所谓“差分”就就是指原子组成体系(团簇)之后电荷得重新分布,“二次”就就是指同一个体系化学成分或者几何构型改变之后电荷得重新分布,因此通过这种差分图可以很直观地瞧出体系中个原子得成键情况。通过电荷聚集(accumulation)/损失(depl etion)得具体空间分布,瞧成键得极性强弱;通过某格点附近得电荷分布形状判断成键得轨道(这个主要就就是对d轨道得分析,对于s或者p轨道得形状分析我还没有见过)。分析总电荷密度图得方法类似,不过相对而言,这种图所携带得信息量较小。?能带结构分析现在在各个领域得第一原理计算工作中用得非常普遍了。但就就是因为能带这个概念本身得抽象性,对于能带得分析就就是让初学者最感头痛得地方。关于能带理论本身,我在这篇文章中不想涉及,这里只考虑已得到得能带,如何能从里面瞧出有用得信息。首先当然可以瞧出这个体系就就是金属、半导体还就就是绝缘体。判断得标准就就是瞧费米能级与导带(也即在高对称点附近近似成开口向上得抛物线形状得能带)就就是否相交,若相交,则为金属,否则为半导体或者绝缘体。对于本征半导体,还可以瞧出就就是直接能隙还就就是间接能隙:如果导带得最低点与价带得最高点在同一个k点处,则为直接能隙,否则为间接能隙。在具体工作中,情况要复杂得多,而且各种领域中感兴趣得方面彼此相差很大,分析不可能像上述分析一样直观与普适。不过仍然可以总结出一些经验性得规律来。主要有以下几点: 1) 因为目前得计算大多采用超单胞(supercell)得形式,在一个单胞里有几十个原
高一数学知识框架第一章集合与函数概念
第二章基本初等函数(I)
必修二立体几何 第一章空间几何体知识结构如下 画三视图的原则:长对齐、高对齐、宽相等 直观图:斜二测画法 斜二测画法的步骤: (1).平行于坐标轴的线依然平行于坐标轴; (2).平行于y轴的线长度变半,平行于x,z轴的线长度不变; (3).画法要写好。 用斜二测画法画出长方体的步骤:(1)画轴(2)画底面 (3)画侧棱(4)成图
第二章 点、直线、平面之间的位置关系 知识结构如下 第三章 直线与方程 从代数表示到几何直观(通过方程研究几何性质和度量) 直线的倾斜角概念:当直线l 与x 轴相交时, 取 x 轴作为基准 , x 轴正向与直线l 向上方向之间所成的角α叫做直线l 的倾斜角 .特别地,当直线l 与x 轴平行或重合时, 规定α= 0° 1 平面含义:平面是无限延展的 2 平面的画法及表示 (1)平面的画法:水平放置的平面通常画成一个平行四边形,锐角画成450,且横边画成邻边的2倍长(如图) (2)平面通常用希腊字母α、β、γ等表示,如平面α、平面β等, 也可以用表示平面的平行四边形的四个顶点或者相对的两个顶点的 大写字母来表示,如平面AC 、平面ABCD 等。 公理1:如果一条直线上的两点在一个平面内,那么这条直线在此平面内 公理1作用:判断直线是否在平面内 公理2:过不在一条直线上的三点,有且只有一 个平面。符号表示为:A 、B 、C 三点不共线 => 有且只有一个平面α,使A ∈α、B ∈α、C ∈α。 公理2作用:确定一个平面的依据。 公理3:如果两个不重合的平面有一个公共点,那么它们有且只有一 条过该点的公共直线。符号表示为:P ∈α∩β =>α∩β=L ,且P ∈L 公理3作用:判定两个平面是否相交的依据 公理4:平行于同一条直线的两条直线互相平行。符号表示为:设a 、b 、c 是三条直线 强调:公理4实质上是说平行具有传递 性,在平面、空间这个性质都适用。 公理4作用:判断空间两条直线平行的依据。 等角定理:空间中如果两个角的两边分别对应平行,那么这两个角相等或互补 直线与平面有三种位置关系: 1)直线在平面内:有无数个公共点 2)直线与平面相交: 有且只有一个公共点 3)直线在平面平行: 没有公共点 平面平行:一个平面内的两条交直线与另一个平面平行,则这两个平面平行 平面互相垂直:一个平面过另一个平面的垂线,则这两个平面垂直 斜率公式: 点到线距离: 平行线距离:
电荷密度图、能带结构、态密度的分析 能带图的横坐标是在模型对称性基础上取的K点。为什么要取K点呢?因为晶体的周期性使得薛定谔方程的解也具有了周期性。按照对称性取K点,可以保证以最小的计算量获得最全的能量特征解。能带图横坐标是K点,其实就是倒格空间中的几何点。纵坐标是能量。那么能带图应该就是表示了研究体系中,各个具有对称性位置的点的能量。我们所得到的体系总能量,应该就是整个体系各个点能量的加和。 主要是从以下三个方面进行定性/定量的讨论: 1、电荷密度图(charge density); 2、能带结构(Energy Band Structure); 3、态密度(Density of States,简称DOS)。 电荷密度图是以图的形式出现在文章中,非常直观,因此对于一般的入门级研究人员来讲不会有任何的疑问。唯一需要注意的就是这种分析的种种衍生形式,比如差分电荷密图(def-ormation charge density)和二次差分图(difference charge density)等等,加自旋极化的工作还可能有自旋极化电荷密度图(spin-polarized charge density)。所谓“差分”是指原子组成体系(团簇)之后电荷的重新分布,“二次”是指同一个体系化学成分或者几何构型改变之后电荷的重新分布,因此通过这种差分图可以很直观地看出体系中个原子的成键情况。通过电荷聚集(accumulation)/损失(depletion)的具体空间分布,看成键的极性强弱;通过某格点附近的电荷分布形状判断成键的轨道(这个主要是对d轨道的分析,对于s或者p轨道的形状分析我还没有见过)。分析总电荷密度图的方法类似,不过相对而言,这种图所携带的信息量较小。 成键前后电荷转移的电荷密度差。此时电荷密度差定义为:delta_RHO = RHO_sc - RHO_atom 其中RHO_sc 为自洽的面电荷密度,而RHO_atom 为相应的非自洽的面电荷密度,是由理想的原子周围电荷分布堆彻得到的,即为原子电荷密度的叠加(a superposition of atomic charge densities)。需要特别注意的,应保持前后两次计算(自洽和非自洽)中的FFT-mesh 一致。因为,只有维数一样,我们才能对两个RHO作相应的矩阵相减。 能带结构分析现在在各个领域的第一原理计算工作中用得非常普遍了。首先当然可以看出这个体系是金属、半导体还是绝缘体。对于本征半导体,还可
图像教程:密度显示 背景 这些图像显示了蛋白数据库 条目2fma 的一部分, Alzheimer's amyloid precursor protein (APP) copper-结合域, 同时它的 电子密度图(2fo-fc) 由电 子密度服务器提供。 图像 How-To 这里的方法仅仅是一个例 子;达到形似的结果通常有很多种路线。也可以参考: presets , tips on preparing images , and Chimera 网站, 中的Image Gallery 和Guide to Volume Display 开启Chimera ,按要求放大窗口。显示命令行(例如,使用Favorites... Command Line )。 从蛋白数据库 取回 2fma 结构,然后应用预设值 #2 显示所有的原子和杂原子color-coding: Command : open 2fma Command : preset apply int 2 从 Electron Density Server 取回此结构的密度图。 Command : open edsID:2fma Click Volume Viewer 对话框的小眼睛图标,先隐藏电子密度图。随时用鼠标按你的要求进行移动和放大操作。 由于一个目的就是显示可替换的构象的例子,通过标签对可替换的构象的残基和原子进行识别。 Command : rlab @.a A: B:
Met-170 和Glu-183有可替换的构象;选择Met-170 附近的一段残基作为图形。只显示残基168-170和其相邻残基的骨架。 Command: ~rlab Command: show :168-170 Command: focus Command: disp :167,171@n,ca,c,o 改变到stick表示方法,使棍更细。 Command: rep stick Command: setattr m stickScale 0.5 只选择侧链Tyr-168 和Met-170; 这次选择将会用于把密度显示限定到一个区域。Command: sel :168,170 & without CA/C1' without CA/C1'部分是进入菜单Select... Structure... side chain/base... without CA/C1'的简洁方法。所有在Select... Structure下的终端菜单可以用作command-line specifiers。 在Volume Viewer中, 1.点击眼睛图标,使密度显示恢复。 2.从菜单选择Features... Zone,点击出现在对话框中的Zone按钮。然后,可以取消 选择(例如,在空区通过ctrl-点击鼠标左键),但是区域半径仍然可以调整。本图中区域半径设置为1.96 ?。 3.柱状图的垂直栏(threshold)代表了轮廓水平; o要改变水平,用鼠标或者键盘在柱状图下面的level区输入不同值拖拽threshold到左边或者右边。 o要改变Color, 点击柱状图下面的正方形color well,使用Color Editor o要添加另一个轮廓水平,Ctrl-点击柱状图; Level 和Color的变化应用最近拖拽或者点击最临界的。 4.从菜单选择Features... Surface and Mesh Options显示额外的显示设置。 Figure A的Volume显示设置(除了默认值) ?Style mesh ?two contour levels: 1.Level 0.426, Color (RGBA) 0.36 1.0 1.0 0.636 2.Level 2.06, Color (RGBA) 1.0 0.0 1.0 1.0 (magenta) ?turned on Smooth mesh lines ?Mesh line thickness 1.5 pixels Figure B的Volume显示设置(除了默认值) ?Style surface ?two contour levels: 1.Level 0.426, Color (RGBA) 0.36 1.0 1.0 0.4 2.Level 2.06, Color (RGBA) 1.0 0.0 1.0 0.6 ?turned on Surface smoothing iterations 2 factor 0.3
【人教版初中数学知识结构图】 1、有理数(正数与负数) 2、数轴 6、有理数的概念3、相反数 4、绝对值 5、有理数从大到小的比较 7、有理数的加法、加法运算律 17、有理数8、有理数的减法 9、有理数的加减混合运算 10、有理数的乘法、乘法运算律 16、有理数的运算11、有理数的除法、倒数 12、有理数的乘方 13、有理数的混合运算 21、代数式14、科学记数法、近似数与有效数字 22、列代数式15、用计算器进行简单的数的运算 23、代数式的值18、单项式 27、整式的加减20、整式的概念19、多项式 24、合并同类项 25、去括号与添括号 26、整式的加减法 28、等式及其基本性质 29、方程和方程的解、解方程 198 32、一元一次方程30、一元一次方程及其解法 初31、一元一次方程的应用33、代入(消元)法 中35、二元一次方程组的解法34、加减(消元)法 数193 36、相关概念及性质 学数39、二元一次方程组37、三元一次方程组及其解法举例 与38、一元方程组的应用40、一元一次不等式及其解法 代45、一元一次不等式43、一元一次不等式41、不等式的解集 数和一元一次不等式组44、一元一次不等式组42、不等式和它的基本性质 46、同底数幂的乘法、单项式的乘法 47、幂的乘方、积的乘方 51、整式的乘法48、单项式与多项式相乘 49、多项式的乘法 56、整式的乘除50、平方差与完全平方公式 52、多项式除以单项式 55、整式的除法53、单项式除以单项式 54、同底数幂的除法 57、提取公因式法 61、方法58、运用公式法 63、因式分解59、分组分解法 62、意义60、其他分解法66、含字母系数的一元 65、分式的乘除法——64、分式的乘除运算一次方程 72、分式69、可化为一元一次方程的分式方程及其应用67、分式方程解法、 70、分式的意义和性质增根 71、分式的加减法68、分式方程的应用 75、数的开方73、平方根与立方根 74、实数 86、二次根式的意义76、最简二次根式 79、二次根式的乘除法77、二次根式的除法
高中数学知识结构图 集合的概念与表示方法 集合集合的性质 集合之间的关系与运算 解析法 函数的概念与表示方法列表法 图像法 定义域 函数的三要素对应关系 值域 单调性 奇偶性 函数的性质周期性 极值 最值一次、二次函数 反比例函数 基本初等函数指数函数与对数函数图像、性质和应用函数函数的分类幂函数 复合函数三角函数 分段函数 函数图像及其变换平移、对称、翻折和伸缩变换 概念 反函数存在条件 与原函数的关系 函数与方程函数的零点对应方程的解 函数的应用建立函数模型 任意角弧度制与三角函数 同角三角函数关系 诱导公式 三角函数中的公式和角、差角公式 二倍角公式与半角公式 三角函数和差化积与积化和差公式 正弦函数三要素 三角函数余弦函数性质 正切函数图像及其变换 正弦定理 解三角形余弦定理 三角形面积
柱体结构 椎体 空间几何体台体三视图和直观图 球体 简单组合体表面积与体积 点、直线、平面的位置关系 点、直线、平面的关系直线、平面平行的性质和判定 直线、平面垂直的性质和判定立体几何点到点的距离 点到直线的距离 空间距离点到平面的距离 直线到平面的距离 平行平面间的距离 异面直线形成的角 空间的角直线与平面形成的角 倾斜角、斜率和截距 点斜式 斜截式 直线直线与方程两点式 截距式 一般式 直线之间的位置关系垂直与平行的条件 圆与方程一般方程与标准方程 几何圆点与圆的位置关系 位置关系直线与圆的位置关系 圆与圆的位置关系 解析几何 圆锥曲线椭圆定义及标准方程 双曲线性质 离心率 点到点的距离 点到直线的距离 平面距离点到圆的距离 两平行线的距离 直线到圆的距离 相离圆的距离 对称问题中心对称关于点对称 轴对称关于直线对称 平面向量概念 向量加减法 向量运算向量的数乘 向量的数量积 空间向量几何意义及应用
§5-7 晶体中电子的能态密度 5.7.1 带底附近的能态密度 在本章第一节中,我们已经得到自由电子的态密度N (E ), 3 212 22()4m N E V E π??= ??? h ………………………………………… …………………………………(5-7-1) 而且N(E)~E 的关系曲线已由图5-7-1给出。晶体中电子受到周期性势场的作用,其能量E(k )与波矢的关系不再是抛物线性质,因此式(5-7-1)不再适用于晶体中电子。下面以紧束缚理论的简立方结构晶格的s 态电子状态为例,分析晶体中电子态密度的知识。 由前面的紧束缚理论,我们已经得到简立方结构晶格的s 能带的E(k )形式为: ()()012cos cos cos s x y z E J J k a k a k a ε=--++k …………………………………………………(5-7-2) 其中能量极小植在Γ点k =(0, 0, 0)处,其能量为()016s E J J ε=--k ,所以在Γ点附近的能量,可以通过将()E k 展开为在k =0处的泰勒级数而得到,以2 cos 12x x =-+L ,取前两项代入,可以得到: ()()()2222222 2011123()2s x y z s x y z E J J a k k k E J a k k k ε??=---++=Γ-++ ??? k …………………(5-7-3) 在第五节,我们已经根据有效质量的定义,算得简立方晶格s 带Γ点处的有效质量为一个标量, 2 21 *02m a J =>h …………………………………………………………………………………………… (5-7-4) 代入后,可得到 ()22 * ()2s k E E m =Γ+h k …………………………………………………………………………………(5-7-5) 式(5-7-5)表明:在能带底k =0附近,等能面是球面,如果以()()s E E -Γk 及* m 分别代替自由电子的能量E 及质量m ,就可得到晶体中电子在能带底附近的能态密度函数: *312 222()4()[()()]s m N E V E E π=-Γh k ……………………………………………………………(5-7-6) 5.7.2 带顶附近的能态密度 能带顶在(,,)a a a πππ=k 的R 点处,容易知道,其能量为()016s E J J ε=-+k 。以R 点附近的 图5-7-1 自由电子能态密度
电压噪声频谱密度(v/sq.rt(Hz)) 运算放大器电路固有噪声的分析与测量 噪声的重要特性之一就是其频谱密度。电压噪声频谱密度是指每平方根赫兹的有效(RMS) 噪声电压(通常单位为nV/sq.rt-Hz)。功率谱密度的单位为W/Hz。在上一篇文章中,我们了解到电阻的热噪声可用方程式 2.1 计算得出。该算式经过修改也可适用于频谱密度。热噪声的重要特性之一就在于频谱密度图较平坦(即所有频率的能量相同)。因此,热噪声有时也称宽带噪声。运算放大器也存在宽带噪声。宽带噪声即:频谱密度图较平坦的噪声。 方程式 2.1:频谱密度——经修改后的热噪声方程式 图2.1:运算放大器噪声频谱密度 除了宽带噪声之外,运算放大器常还有低频噪声区,该区的频谱密度图并不平坦。这种噪声称作1/f噪声,或闪烁噪声,或低频噪声。通常说来,1/f 噪声的功率谱以 1/f 的速率下降。这就是说,电压谱会以1/f(1/2 ) 的速率下降。不过实际上,1/f 函数的指数会略有偏差。图2.1 显示了典型运算放大器在1/f 区及宽带区的频谱情况。请注意,频谱密度图还显示了电流噪声情况(单位为 fA/rt-Hz)。 我们还应注意到另一点重要的情况,即1/f 噪声还能用正态分布曲线表示,因此第一部分中介绍的数学原理仍然适用。图2.2 显示了1/f 噪声的时域情况。请注意,本图的 X 轴单位为秒,随时间发生较慢变化是1/f 噪声的典型特征。
图2.2:时域所对应的 1/f 噪声及统计学分析结果 图2.3 描述了运算放大器噪声的标准模型,其包括两个不相关的电流噪声源与一个电压噪声源,连接于运算放大器的输入端。我们可将电压噪声源视为随时间变化的输入偏移电压分量,而电流噪声源则可视为随时间变化的偏置电流分量。 图2.3:运算放大器的噪声模型 运算放大器噪声分析方法 运算放大器噪声分析方法是根据运放数据表上的数据计算出运放电路峰-峰值输出噪声。在介绍有关方法的时候,我们所用的算式适用于最简单的运算放大器电路。就更复杂的电路而言,这些算式也有助于我们大致了解可预见的噪声输出情况。我们也可针对这些更复杂的电路提供较准确的计算公式,但其中涉及的数学计算将更为复杂。对更复杂的电路而言,或许我们最好应采用三步走的办法。首先,用算式进行粗略的估算;然后,采用 spice 仿真程序进行更准确的估算;最后通过测量来确认结果。 我们将以 TI OPA277 的简单非反向放大器为例来说明有关电路的情况(见图2.4)。我们的目标是测定峰峰值输出噪声。为了实现这一目的,我们应考虑运算放大器的电流噪声、电压噪声以及电阻热噪声。我们将根据产品说明书中的频谱密度曲线来确定上述噪声源的大小。此外,我们还要考虑电路增益与带宽问题。
能带结构和态密度图的绘制及初步分析 前几天在QQ的群中和大家聊天的时候,发现大家对能带结构和态密度比较感兴趣,我做计算已经有一年半了,有一些经验,这里写出来供大家参考参考,希望能够对初学者有所帮助,另外写的这些内容也不可能全都正确,只希望通过表达出来和大家进行交流,共同提高。 MS这个软件的功能确实是比较强,但是也有一些地方不尽如人意的地方。(也可能是我对一些结果不会分析所致,有些暂时不能解决的问题在最后一部分提出,希望大家来研究 研究,看看有没有实现的可能性)。 能带结构、态密度和布居分析是很重要的内容,在 分析能带结构和态密度的时候,往往是先作图,然后分 析。 软件本身提供的作图功能并不是很强,比如说能带结构 (只能带只能做point图和line图),不美观不说,对于 每一个能带的走势也不好观察,感觉无从下手。所以我 一般用origin作图(右图是用origin做的能带图)。能带 结构和态密度的作图过程请参考我给大家提供的动画。 接下来我们先开看看能带结构的分析和制作! 第一部分:能带结构 这个部分打算先简单的介绍一下能带的基础知识,希望能对大家有所帮助,如果对能带了解比较深入的朋友,可以跳过这个部分内容,之中不当之处请勿见笑。^_^ 第一个问题是: 1、能带是怎样形成——轨道和一维体系的能带。 这是最基本的一个问题,我们要对能带结构进行分析,首先要知道它是如何来的。其实能带是一种近似的结果(可以看成一种近似),是周期边界条件(bloch函数)下的一种近似。先来看看一个最简单的问题,非周期体系有没有能带结构?答案是没有的,大家可以试试: ①建一个周期的晶胞②选择build菜单下的symmetry子菜单下的none periodic superstructure去掉周期边界条件性③看看还能够运行吗?运行(run)按钮变灰了,不能提交作业了。这说明什么问题?这说明这个CASTEP这个模块不能计算非周期的体系,另外可以参考MS中的DMOL模块,它可以计算非周期系统,虽然可以计算周期系统,但是仍不能计算能带,大家可以试试,看看property中的band structure能不能选上,一定不能!!^_^ 从这里,我们可以得到一个结论,对于单个原子(分子、单胞)如果不加上周期边界条件,是无法获得能带结构的。所以计算小分子体系,或者采用团簇模型的朋友,这部分内容或许对你们没有帮助!那么,非周期体系的态密度能够计算吗?这应该是能够计算的,曾经开到过文献采用团簇模型,计算出态密度的(phys. Rev. 上的文章)。 那么非周期体系为什么没有能带结构呢? 看一个例子:一个H2分子有能带吗?没有,因为它没有周期边界条件,也就是说在x,y,z方向上没有重复,所以它没有能带结构。那H2分子有什么东西呢?有两个轨道,两个 1s原子轨道,或者说两个轨道能级,它们成键参考右图。 再看另外一个例子:一维无限H原子链 H H H H H H 在一维无限H原子链体系中,产生了能带。 为什么在一维无限H原子链体系中能够产生能带呢?
高中数学必修知识框架图
{1.11.2 1.32.12.22.33.1?????????????????????????集合第一章:集合与函数概念 函数及其表示函数的基本性质指数函数必修一第二章:基本初等函数(Ⅰ)对数函数幂函数第三章:函数的应用 函数与方程1.11.21.32.12.22.33.13.23.3???????????????空间几何体的结构第一章:空间几何体 空间几何体的三视图和直观图空间几何体的表面积与体积空间点、直线、平面之间的位置关系第二章:点线面的位置关系直线、平面平行的判定及其性质直线、平面垂直的判定及其性质必修二直线的倾斜角与斜率第三章:直线与方程 直线的方程直线的交点坐标与距离第四章:圆与方程 4.14.24.3?????????????????????????? 圆的方程 直线、圆的位置关系空间之间坐标系1.11.21.32.12.22.33.13.23.3???????????????????????????????? 算法与程序框图第一章:算法初步基本算法语句算法案例随机抽样必修三第二章:统计 用样本估计总体 变量间的相关关系随机事件的概率第三章:概率 古典概型 几何概型
1.11.21.31.41.5)1.6 2.12.22.32.42.5x ω?????????+????????????任意角和弧度制任意角的三角函数三角函数的诱导公式第一章:三角函数 三角函数的图像与性质函数y=Asin(的图像三角函数模型的简单应用平面向量的实际背景及基本概念必修四平面向量的线性运算第二章:平面向量 平面向量的基本定理及坐标表示平面向量的数量积平面向量应用举例第 3.13.2?????????????????????????? 两角和与差的正弦、余弦和正切公式三章:三角恒等变换 简单的三角恒等变换1.11.22.12.22.3n 2.42.5n 3.13.23.33.42a b ab ????????????????????????????????+?≤??正弦定理和余弦定理第一章:解三角形应用举例数列的概念与简单表示法等差数列第二章:数列 等差数列的前项和必修五等比数列等比数列的前项和不等关系与不等式一元二次不等式及其解法第三章:不等式 二元一次不等式(组)与简单的线性规划问题基本不等式:???????????
如何分析第一原理的计算结果 用第一原理计算软件开展的工作,分析结果主要是从以下三个方面进行定性/定量的讨论: 1、电荷密度图(charge density); 2、能带结构(Energy Band Structure); 3、态密度(Den sity of States,简称DOS)。 电荷密度图是以图的形式出现在文章中,非常直观,因此对于一般的入门级研究人员来讲不会有任何的疑问。唯一需要注意的就是这种分析的种种衍生形式,比如差分电荷密图(d ef-ormation charge density)和二次差分图(differenee charge density)等等,加自旋极化的工作还可能有自旋极化电荷密度图(spin-polarized charge density)。所谓差分”是指原子组成体系(团簇)之后电荷的重新分布,二次”是指同一个体系化学成分或者几何构型 改变之后电荷的重新分布,因此通过这种差分图可以很直观地看出体系中个原子的成键情况。通过电荷聚集(accumulation)/损失(depletion )的具体空间分布,看成键的极性强弱;通过某格点附近的电荷分布形状判断成键的轨道(这个主要是对d轨道的分析,对于s 或者p轨道的形状分析我还没有见过)。分析总电荷密度图的方法类似,不过相对而言,这种图所携带的信息量较小。 能带结构分析现在在各个领域的第一原理计算工作中用得非常普遍了。但是因为能带这个概念本身的抽象性,对于能带的分析是让初学者最感头痛的地方。关于能带理论本身,我在这篇文章中不想涉及,这里只考虑已得到的能带,如何能从里面看出有用的信息。首先当然可以看出这个体系是金属、半导体还是绝缘体。判断的标准是看费米能级和导带(也即在高对称点附近近似成开口向上的抛物线形状的能带)是否相交,若相交,则为金属,否则
态密度计算 态密度:表示单位能量范围内所允许的电子数,也就是说电子在某一能量范围的分布情况。因为原子轨道主要是以能量的高低去划分的,所以态密度图能反映出电子在各个轨道的分布情况,反映出原子与原子之间的相互作用情况,并且还可以揭示化学键的信息。态密度有分波态密度(PDOS)和总态密度(TDOS)形式。 原则上讲,态密度可以作为能带结构的一个可视化结果。很多分析和能带的分析结果可以一一对应,很多术语也和能带分析相通。但是因为它更直观,因此在结果讨论中用得比能带分析更广泛一些。 计算过程:主要分成三步:一、结构优化;二、静态自洽计算;三、非自洽计算。 1,结构优化:原子弛豫,确定体系内每个原子位置。常用INCAR。2,静态自洽计算:(得到自洽的电荷密度CHG、CHGCAR和E-fermi,提供给下一步非自洽计算用) INCAR设置注意,ICHARG = 2 3,非自洽计算(准确计算电荷分布) INCAR设置:ISTART=1(若存在WAVECAR文件时取1);ICHARG=11(表示从CHGCAR中读入电荷分布,并且在计算中保持不变);RWIGS (或LORBIT=11(或10),这时可不设RWIGS); 计算完成时,生成DOSCAR,采用spit_dos.dl小程序把dos分开(注意vp.dl要拷到同目录下),会生成N+1个文件,DOS0为总态密度,DOS1到DOSN为N个原子的分态密度。每个分态密度共7列分布为
—能量→Sup→Sdown→Pup→Pdown→Dup→Ddown 不知道从态密度能否定性分析出来,因为态密度越尖,则电子的局域性越强, 修正版的splitdos有三个文件:vp、sumdos和split_dos.ksh INCAR设置: ISTART = 1;ICHARG = 11 LORBIT = 10 【对于PAW势,可设置LORBIT = 10,此时可不用设置RWIGS参数】或者设置RWIGS参照POTCAR