工艺技术风险管理表
1.1 风险分析标准
从风险的严重性、频率数和检测性三方面对工艺存在的风险进行分析评估,判定标准如下:1.1.1 严重性分级标准
1.1.2 频率分级标准
1.1.3 可检测性分级标准
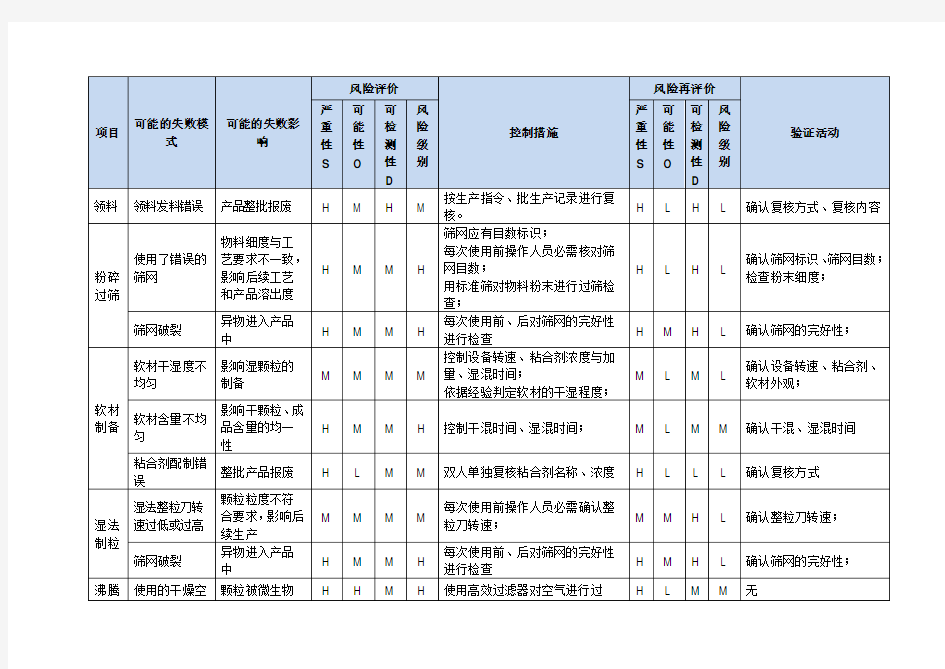
1.2 风险分析列表
ISPE-CCPIE CHINA CONFERENCE 2012
工艺风险评估关键质量属性&关键工艺参数
September 24-25 2012 Beijing
1
目录
? 为什么必须要找到CQA&CPP? ? 如何定义CQA&CPP? ? 如何开展CQA&CPP风险评估工作? ? ISPE 知识概述
ISPE-CCPIE CHINA CONFERENCE 2012
2
术语
? CQA(Critical Quality Attribute)关键 质量属性
? CPP(Critical Process Parameter) 关 键工艺参数
ISPE-CCPIE CHINA CONFERENCE 2012
3
为什么必须要找到CQA&CPP?
ISPE-CCPIE CHINA CONFERENCE 2012
4
为什么必须找到CQA&CPP?
? 法规规定 ? 指南要求
ISPE-CCPIE CHINA CONFERENCE 2012
5
SFDA GMP2010年版
? 第二章 质量管理 第四节质量风险管理
? 第十三条 质量风险管理是在整个产品生命周期 中采用前瞻或回顾的方式,对质量风险进行评估 、控制、沟通、审核的系统过程。
? 第十四条 应当根据科学知识及经验对质量风险 进行评估,以保证产品质量。
? 第十五条 质量风险管理过程所采用的方法、措 施、形式及形成的文件应当与存在风险的级别相 适应。
ISPE-CCPIE CHINA CONFERENCE 2012
6
1.0 范围 适用于公司生产制造过程中影响产品交付和产品质量的风险识别和控制因素策划及实施。 2.0 目的 为了控制可能会影响产品交付和产品质量有关的风险,为风险识别、评估和降低 确定技术、工具和对其应用,特制定本程序。 3.0 设备要求 不适用 4.0 职责 4.1 设备部负责对生产过程中产生质量风险、检验文件的制定和更改以及设 备设施的可利用性、可维护性的风险进行评估和控制; 4.2 营销主料采购和供应链辅料采购部负责对原辅材料采购、外包方和供方 业绩进行评估和控制;负责供方变化及其质量管理体系变化产生的风险 进行评估和控制; 4.3 销售/客服部和工程部负责不合格产品交付产生的风险进行评估和控制 4.4 质量安全部负责产品的检验与试验过程、检测设备的使用和维修 可能产生的质量风险进行评估和控制; 4.5 人事行政部负责对人员能力、专项技能的符合性、组织机构变更以及关 键或重要人员的改变产生的风险进行评估和风险控制。 5.0 程序 5.1 定义 5.1.1风险:有可能发生并有潜在负面结果的局面或境况 5.1.2 风险评估 评估来自可能发生的风险,考虑现有控制的适当性和决定该风险是否可接 受的过程。 5.2 风险辨识和风险评估过程 1)评估准备----收集资料,制定计划 2)风险识别----事故类型,影响因数及机制 3)风险评估----事故发生的可能性,事故的严重程度,风险值的确定,风险分级 4)风险控制----制定方案,落实风险防范措施 5)登记重大风险项目 6)定期检查提出整改方案 5.3 风险源的识别 5.3.1 风险源的识别应考虑以下方面: 5.3.1.1与产品交付相关的风险评估应包括: 1)设施/设备的可用性和可维护性 2)供应商绩效以及材料的可用性/供应
1 目的 建立片剂制剂工艺质量风险评估,确认工艺参数、操作对产品质量的影响。 2 适用范围 片剂各品种的制剂各生产工序。 3 责任 相关人员各负其责 4 内容 4.1 风险评估小组组成 4.2 概述 4.2.1 本风险评估将按照片剂工艺流程展开评估: 配料制粒整粒压片包衣包装 配料的目的是按工艺要求将药物的各个有效成分与粘合剂、崩解剂等辅料经过处理后混合在一起,使其达到基本均匀、性状相对一致,便于后工序的生产,保证产品的质量;制粒是将物料由粉末状改变为颗粒状的过程,制得颗粒需具有良好的流动性和可压性,同时须具有一定的密度;整粒是将颗粒和工艺处方中剩余的物料经过处理后混合在一起,使其达到粒度均匀、含量均匀、性状一致,便于后工序的生产,保证产品的质量;压片是将颗粒压制成片剂,目的是赋予药物一定的形状和对药物进行分剂量,也是便于后工序的生产,保证产品的质量;包衣是在素片的外面包上一层衣,主要目的是使药物便于储存,保证药物性质稳定,保证产品质量;包装的目的是使成品便于分发、应用和贮藏,保证产品在有效期内的质量。整个生产流程对产品质量都很关键,所以需对工艺各个流程进行工艺质量风险评估,经评估便于有潜在风险的工艺采取相应的措施,从而保证产品质量。 4.2.2 依据SMP-QA-048-00采用失败模式对固体制剂各工序可能的风险进行定量分析评估。 4.2.2.1 将风险的等级用定量的方式来描述: 风险严重性(S)评估表
2 风险发生可能性(P)评估表风险可发现性(D)评估表 RPN(定量分级值)=S×P×D。当RPN值为≤16时,风险等级定为低;16 生产工艺质量风险评估 严重性分级标准高(H)失败使中间产品或成品质量指标出现不合格或产生质量隐患,或对批产品质量均一性产生不良影响,导致该批产品全部做报废处理。中(M)失败使中间产品某项质量指标不合格,但可经偏差处理,且不会对批产品质量均一性产生不良影响,不会导致成品有什么质量缺陷或隐患。低(L)失败只会造成个别产品质量指标不合格,但可以百分之百将不合格品剔除。1、1、2 频率分级标准高(H)发生可能性每季高于1次。中(M)发生可能性每年高于1次,低于4次。低(L)发生可能性每年低于1次。1、1、3 可检测性分级标准高(H)具有反映质量均一性的质量指标,如含量均匀度、有关物质等能被仪器、设备100%控制和检测。中间产品质量与成品质量指标均对同一质量项目进行控制和检测。中(M)涉及水分、装量等能被仪器、人员进行离线检测,但如不对过程进行有效控制,产品质量均一性无法确认,检测结果应被怀疑的质量指标。低(L)无法对影响产品质量的控制指标的真实性进行有效监控,因此可能会影响到产品质量均一性,也可能会造成整批产品报废,可通过产品质量间接反映出问题。1、2 风险分析列表项目可能的失败模式可能的失败影响风险评价控制措施风险再评价验证活动严重性S可能性O可检测性D风险级别严重性S可能性O可检测性D风险级别领料领料发料错误产品整批报废HMHM按生产指令、批生产记录进行复核。 HLHL确认复核方式、复核内容粉碎过筛使用了错误的筛网物料细度与工艺要求不一致,影响后续工艺和产品溶出度HMMH筛网应有目数标识;每次使用前操作人员必需核对筛网目数;用标准筛对物料粉末进行过筛检查;HLHL确认筛网标识、筛网目数;检查粉末细度;筛网破裂异物进入产品中HMMH每次使用前、后对筛网的完好性进行检查HMHL确认筛网的完好性;软材制备软材干湿度不均匀影响湿颗粒的制备MMMM控制设备转速、粘合剂浓度与加量、湿混时间;依据经验判定软材的干湿程度;MLML确认设备转速、粘合剂、软材外观;软材含量不均匀影响干颗粒、成品含量的均一性HMMH控制干混时间、湿混时间;MLMM确认干混、湿混时间粘合剂配制错误整批产品报废HLMM双人单独复核粘合剂名称、浓度HLLL确认复核方式湿法制粒湿法整粒刀转速过低或过高颗粒粒度不符合要求,影响后续生产MMMM每次使用前操作人员必需确认整粒刀转速;MMHL确认整粒刀转速;筛网破裂异物进入产品中HMMH 每次使用前、后对筛网的完好性进行检查HMHL确认筛网的完好性;沸腾干燥使用的干燥空气不洁净颗粒被微生物污染HHMH使用高效过滤器对空气进行过滤,并定期维护保养、更换HLMM无使用的干燥空气风量不足延长干燥时间,影响药品水分含量MMMM控制风机频率,确保风量;控制抖袋频次,防止堵塞;MLML确认风机频率、抖袋频次;加热温度不足延长干燥时间,影响药品水分含量MMMM控制进风温度、物料温度及干燥时间; MLML确认进风温度、物料温度及干燥时间;干燥颗粒损耗大物料平衡、收率不合 2011年11月 1 2011年11月 2 Figure 28. Process map for Example MR Tablets, 10 mg 图28 实例10 mgMR片剂的工艺图 2011年11月 3 Example QbD MR Tablet Module 3 Quality 3.2.P.2 Pharmaceutical Development 实例QbD MR片剂模块3质量3.2.P.2药物开发 2.3.1 Initial Risk Assessment of the Drug Product Manufacturing Process 药品生产工艺的初始风险评估 A risk assessment of the overall drug product manufacturing process was performed to identify the high risk steps that could affect the final drug product CQAs. Subsequently, the drug product intermediate CQAs that are directly linked to the identified final drug product CQAs were identified. The process variables that could impact the identified drug product intermediate CQAs became the focus of the risk assessment to determine which variables have the highest potential to cause a CQA failure. These variables then needed to be investigated in order to optimize the drug product manufacturing process and reduce the risk of failure. For example, the overall risk assessment of the manufacturing process found assay of the tablets to be at high risk of failure due to the drug layering step. Subsequently, assay of the layered beads is directly linked to final tablet assay and was identified as the CQA of the drug-layered beads. Process variables that could directly impact the assay of the drug-layered beads were assessed to identify which of the variables could have the highest potential to cause a bead assay failure. This method of identifying process variables for further study was also illustrated previously in Figure 19. 进行整体药品生产工艺的风险评估以确定可能影响最终药品CQAs的高风险步骤。随后,确定直接与已确定的最终药品CQAs相关的药品中间体CQAs。可能影响已确定的药品中间体CQAs的工艺变量成为风险评估的焦点,以确定哪些变量最可能引起CQA不合格。然后需要研究这些变量以便优化药品生产工艺并降低不合格的风险。例如,整体生产工艺的风险评估发现片剂的含量是不合格的高风险由于层积上药步骤。随后,层积微丸的含量直接与最终片剂含量相关,曾被确定为层积上药微丸的CQA。评估可直接影响层积上药微丸含量的工艺变量以确定哪些变量最有可能引发微丸含量不合格。这种确定用于进一步研究的工艺变量的方法之前也在图19中进行了说明。 In the initial risk assessment of the overall manufacturing process shown in Table 51, drug layering, ER polymer coating, blending and lubrication, and compression were identified as high risk steps that could affect the quality of the final product. Justification for each risk assignment is presented in Table 52. 表51所示的整体生产工艺的初始风险评估中,确定层积上药,ER聚合物包衣,混合和润滑,及压缩为可影响最终药品质量的高风险步骤。每个风险分配的依据见表52。 复方氨酚那敏颗粒 质量风险评估报告 报告起草:年月日报告审核:年月日报告批准:年月日 有限公司 二0一三年八月 目录 1.复方氨酚那敏颗粒注册相关信息 (1) 2.复方氨酚那敏颗粒质量风险概述 (1) 3.复方氨酚那敏颗粒质量风险识别 (1) 4.风险分析 (3) 4.1复方氨酚那敏颗粒风险失败模式建立 (3) 4.2复方氨酚那敏颗粒风险分析 (4) 4.2.1人员风险 (4) 4.2.2设备、仪器风险 (5) 4.2.3复方氨酚那敏颗粒用物料风险 (6) 4.2.4复方氨酚那敏颗粒生产操作方法风险 (7) 4.2.5复方氨酚那敏颗粒生产环境风险 (10) 4.2.6复方氨酚那敏颗粒检验(测量)风险 (13) 5.评估总结论与建议 (14) 6.本风险评估依据与资料收集范围 (15) 产品名称:复方氨酚那敏颗粒产品阶段:生产全过程 评估小组成员: 评估日期: 1.复方氨酚那敏颗粒注册相关信息 药品注册基本信息:通用名称:复方氨酚那敏颗粒,规格:每袋装10g,有效期:24个月。批准注册认证信息:取得批件时间:,批件有效期:5年,再注册时间:,批准文号:国药准字,执行标准:国家食品药品监督管理局标准WS-10001-(HD-0256)-2002。 我公司复方氨酚那敏颗粒的工艺规程、质量标准以及包装标签上的信息均按上述信息执行,与注册信息相符合。 2.复方氨酚那敏颗粒质量风险概述 本报告复方氨酚那敏颗粒质量风险进行系统的分析评估,对复方氨酚那敏颗粒涉及的生产过程所有可能出现的风险进行评估,确定重点控制的目标,制定纠正和预防措施,对于高风险和中等风险的没有管理措施的必须确定降低风险的措施,低风险加强生产过程控制,确保产品质量,降低风险发生的可能性,提高可识别性,将风险控制在可接受水平。如果采取风险控制措施和预防措施后风险仍不可接受,应重新制定降低风险的措施和办法。本风险评估资料来源于公司复方氨酚那敏颗粒质量档案,历年生产记录,各种涉及的偏差、变更、验证、工艺规程、质量标准涉及等复方氨酚那敏颗粒生产的全部记录资料。 3、复方氨酚那敏颗粒风险识别 复方氨酚那敏颗粒质量风险的识别用鱼骨图来描述,根据鱼骨图(见下图)逐一展开分析和评价,找出风险点进行控制,把降低风险的措施落实到每个环节。 ...............有限公司片剂工艺验证方案文件编号JM-JSB-ZYZ(01)-053-02 ...............有限公司 生产质量技术标准文件 .....片工艺验证方案 文件编号:JM-JSB-ZYZ(01)-053-02 2020年03月 ...............有限公司 验证立项申请表 ...............有限公司 验证方案审批表 验证小组成员及职责 验证实施前对参与验证人员进行确认,小组成员经过本方案的培训,掌握验证目的和验证程序及可接受标准。 目录 1.验证目的 2.验证范围 3.产品概况 4.验证涉及文件 5.验证背景 6.片剂生产工艺质量风险评估 7.工艺验证方法及验证实施 7.1验证要求 7.2验证实施 7.2.1药粉制备工序验证实施 7.2.2提取浓缩收膏工序验证实施 7.2.3喷雾制粒干燥工序验证实施 7.2.4整粒、总混工序验证实施 7.2.5压片工序验证实施 7.2.6包糖衣工序验证实施 7.2.7铝塑包装工序验证实施 7.3收率或物料平衡 7.4成品质量的检测 7.5片剂持续稳定性考察 8.偏差及变更 9.验证时间安排 10.拟定再验证周期 11.评价报告及验证结论 12.验证证书 13.培训 附件1:验证方案修改申请及批准书 片剂工艺验证方案 1.验证目的 工艺验证是在预先经过确认所有的生产条件下,确认按规定的生产工艺规程生产的最终产品符合质量标准的要求。确认该工艺是有效的、可重现的。 通过对生产过程各工序的质量控制指标的检测及各个岗位的操作数据分析,对片剂生产照预先设定的目标和指标进行对比,对过程工艺执行情况进行综合的评价和系统的分析,综合评定本生产线生产工艺的安全性、稳定性和可靠性。 通过验证可以提前发现问题,确保生产出符合预定用途和注册要求的产品。所以对片剂生产线进行全面的工艺验证。本方案是以连续生产三批片剂[批量为150万片(525kg)]为代表,对片剂生产工艺进行验证。 验证过程严格按本方案规定的内容进行,若因特殊原因确需修改时,应填写“验证方案修改申请及批准书”(附件1),报验证委员会批准。 2.验证范围 本验证方案适用于在本方案指定的厂房、设施、设备、工艺条件下片剂的生产工艺验证,当上述条件改变时,应重新验证。 3.产品概况 3.1品名:片剂 3.2剂型:片剂 3.3性状:本品为糖衣片,除去糖衣后显褐色;味微苦涩。 3.4规格:基片重0.35克 3.5处方和依据 3.5.2依据:国家食品药品监督管理局标准YBZ03012008 3.6片剂药材提取、粉碎工艺流程图 【经典资料,WORD文档,可编辑修改】 【经典考试资料,答案附后,看后必过,WORD文档,可修改】 XXX制药有限公司 质量风险评估报告 风险项目:纯化水系统风险评估 编号: 起草人: 年月日审核人: 年月日批准人: 年月日 目录 1.概述 2.目的 3.相关法规指南和参考文献 4.质量风险管理小组人员及其职责分工 5.风险识别 6.风险分析及评价标准 7.风险评估结果及控制 8.风险管理评审结论 9.风险评估报告审批 1. 概述 我公司纯化水系统采用二级反渗透法制备。反渗透是依靠反渗透膜在比自然渗透压力更大的压力下,使水通过反渗透膜变成纯化水,从而达到除去水中盐分的目的。使其制备的水质符合2010版《中国药典》纯化水的质量标准。 1.1系统基本情况 项目 技术参数 工作压力 10-17bar 原水压力 进预处理 2-4bar 进R.O 设备 1.4-3.4bar 一级系统工作压力 10-17bar 二级系统工作压力 10-17bar 1.2 制水工作流程图 2.目的 2.1为降低和控制车间纯化水系统相关的风险,建立有效的纯化水系统质量控制体系,提高产品质量提供风险分析参考。 2.2为车间纯化水系统的验证确认活动提供风险分析参考。 2.3为车间纯化水系统日常运行和产品的质量控制提供风险分析参考。 3. 相关法规指南和参考文献 3.1 《药品生产质量管理规范》(2010年修订) 3.2 2010版GMP 实施指南 原水箱 原水增压泵 多介质过滤器 活性碳过滤器 紫外消毒 一级RO 系统 中间水箱 二级RO 系统 纯化水箱 纯水泵 软水器 精密过滤器 用水点 XXXXX原料药工艺风险评估(样本) 一、风险等级评估 风险等级严重性可能性可检测性 对关键质量属性有影响操作范围接近设计空间或注风险发生及有 高必须严格控制才能保证册范围或参数范围较窄,参发生趋势时可以质量参数偏离范围为关数本身较难控制,正常情况立即被发现。 键性偏差。下可能会偏离范围。 对关键质量属性可能有操作范围接近设计空间或注发现发生后稍 中影响,不严格控制会出册范围或参数范围较宽,参后才能被发现 现质量大的偏差数本身容易控制,异常情况 下才会偏离范围。 对关键质量属性影响很操作范围远比设计空间或注发现发生后很 低小,参数偏离范围为小册范围窄或参数范围比较宽,久后才能被发现偏差或微偏差紧急情况下才会偏离设计空 间 二、风险评估矩阵 1、风险等级矩阵 2、内在关键性矩阵 高高高高严高潜在关键关键 严 中低中高重中非关键潜在关键 重 低低低中性低非关键非关键潜在 性低低中高低中高 可能性可能性 3、可检测性和内在关键性矩阵 4、评估相关性报告文件 最终关键性序名称描述 可高非关键非关键关键号 1 产品属性评估表影响工艺步骤评估 检中非关键关键关键 2 产品质量属性评估表影响工艺参数评估 3 产品质量属性评估表工艺参数关键性评估 测低非关键关键关键 4 产品质量属性评估表工艺参数关键性评估 性非关键潜在关键(采取控制措施后)内在关键性 三:产品工艺风险评估 1、风险评估小组确定评估小组负责人,成员有研发专家、技术转移人员、生产操 作人员、工程人员、项目人员、验证人员、QA、QC、供应商(如必要); 2、寻找产品质量属性(CQA)和关键工艺参数(CPP)的必要前提条件: 1)文件资源: A 保证评估前已具备所有必要的文件; B 详细的文件资料能有效支持CQA和CPP风险评估的执行,有资料需求单。 2)良好培训 A GMP知识培训; B 质量风险管理(ICHQ9); C 公司质量风险管理规程; D 产品工艺知识; E 产品质量属性和关键工艺参数评估程序; F 产品质量属性和关键工艺参数风险评估模板。 3)评估会议 A 风险评估小组成员的资质和业务能力; B 风险评估的主题; C 细致的风险评估记录。 4)风险评估工具失效模式分析(FMEA)。 3、名词定义 1)产品质量属性和关键工艺参数:原料药产品→原料药关键质量属性→潜在关键参数→关键参数→关键步骤→其他控制点 2)产品质量属性:如鉴别、物化性质、外观性状、含量、纯度、粒度、晶型、微生物限度(如需要)。 3)关键工艺参数:工艺设计空间范围或注册参数范围,范围按宽、窄划分。 4)操作参数:指生产过程中可直接控制的输入变量或条件,通常这些参数是物理或化学参数,如温度、加工时间、柱流速、柱清洗体积或浓度、缓冲液、pH对。也称工艺参数。 5)关键操作参数:指一个输入的工艺参数,应控制在有意义、较窄的操作范围的以保证药物成分的质量属性符合要求。尽管较宽运行范围的参数也可能影响产品质量,但一般这些参数比较容易控制,而不易出现影响产品质量偏离,所以有较低的风险。与关键工艺参数为同义词。 6)非关键操作参数:关键运行参数以外所有输入工艺参数,非关键操作参数分为关键操作步骤和非关键操作步骤。 7)重要操作参数:应谨慎的控制在狭窄的范围内是工艺性能必不可少的输入工艺参数,关键操作参数不会影响产品质量关键产品属性,如果超过规定范围则可能影响工艺(如收率、持续时间等),但不会影响到产品质量。 8)非重要操作参数:经过证明的容易控制或有较宽接受限度的输入工艺参数。如果超出非关键操作参数的运行范围,可能对药物成分的质量或工艺性能有影响。 9)设定点:一个操作参数的目标值,设定点范围通常需要在生产规程或批记录中确定。 10)操作单元:一个在生产工艺中独立的步骤或操作,工艺和操作参与被定义以实现一个特殊的工艺目标。也称操作步骤。 4、产品质量属性的判定 药品生产过程质量风险评估报告模板 XXXX胶囊生产过程质量风险管理报告 起草人:审核人:审核人:批准人:质量风险管理号:QRM- 起草日期:审核日期:审核日期:批准日期: XXXX药业有限公司 年月年月年月年月 日日日日 目录 XXXX胶囊生产过程质量风险管理报告 1、简介 2、目的 3、范围 4、引用资料 5、风险管理小组组员及职责分工 6、质量风险管理流程 7、风险管理过程 8、风险管理总结及结论 9、风险管理回顾审核 XXXX胶囊生产过程质量风险管理报告 1.简介: 1.1产品概述:XXXX胶囊为以化学原料药XXXX和适量等辅料制成的化学药胶囊剂制剂,为耳鼻喉科及皮肤科用药类非处方药药品。用于缓解过敏性鼻炎有关的症状,如喷嚏、流涕、鼻痒、鼻塞以及眼部痒及灼烧感。口服药物后,鼻和眼部症状及体征得以迅速缓解。亦适用于缓解慢性荨麻疹、瘙痒性皮肤病及其他过敏性皮肤病的症状和体征。规格为10 毫克;贮藏:遮光,密闭保存;包装:铝塑泡罩包装。每板6粒,每盒1板;每板6粒,每盒2板;每板12粒,每盒1板,每板4粒,每盒1板。有效期:30个月。 1.2生产批量:35万粒,140万粒。 1.3主要生产工艺过程及参数: ,混合速度900转/分,混合时间20分钟;批量为140万粒的预混合和35万粒相同,但原辅料均分4等分进行4次混合。 1.3.3 粘合剂15%聚维酮K30的乙醇溶液的配制:按聚维酮K30:95%乙醇量=1.5:8.5(重 量比)进行配置,溶解完全100目滤布过滤。 ,用GHL-250型高效混合制粒机中(每次35万粒量),设定混合I和切割I开动设备混合10分钟后,徐徐加入粘合剂(15%聚维酮K30的乙醇溶液),加完后继续混合3-4分钟,收集软材。 ,筛网目数为24目。 ,每次干燥量为35万粒胶囊的颗粒量,控制干燥温度为50℃~70℃,干燥时间20分钟。,筛网目数为24目。 ,混合速度900转/分,混合时间20分钟;批量为140万粒的总混:用EYH-1000型二维运动混合机进行,混合时间35分钟(此设备无速度调节)。 ,二号胶囊充填模具,用二号空心胶囊进行充填,装量差异控制在充填量的±8%范围内,充填速度为800~1200粒/分。充填过程中,每20分钟检查一次装量。将充填好的胶囊用HTP-Ⅱ型胶囊、片剂抛光机抛光。 ,同时产品冲印批号、有效期。按4粒/板、6粒/板或12粒/板泡罩规格采用相应的二号胶囊泡罩模具,上、下封温度约为120℃,热封温度为160℃。 ,每个盒子及纸箱均打印产品批号、生产日期、有效期,电子监管码?;按6粒/板×1板/盒×200盒/箱或12粒/板×1板/盒×200盒/箱或6粒/板×2板/盒×200盒/箱或4粒/板×1板/盒×200盒/箱的包装规格进行包装。 1.4中间产品的贮存要求:XXXX胶囊颗粒(总混后)用洁净塑料袋盛装,扎紧袋口置洁净的不锈钢桶中密闭,在口服制剂车间中间站贮存,贮存期:天;XXXX胶囊(批,产品均符合标准规定,未产生过质量事故,也发生过严重的毒副作用及不良反应。 2、目的: 2.1通过对XXXX胶囊生产过程质量风险管理,对其带来的质量风险进行有效控制,保证产品质量。 面进行风险梳理识别。 (1)风险严重程度、发生几率、发现的可能性评分标准及风险级别评判标准及风险控制措施:见下表1. 表2. 表3.表4. 液体制剂生产过程质量风险评估报告 目录 1.概述 2. 目的 3.风险管理人员及其职责分工 4.风险分析 5.风险评估 6.风险管理评审结论 1.概述 我公司液体制剂车间主要生产10ml 和100ml 中药合剂,为确保液体制剂车间能顺利的生产出符合标准和规范要求的产品(合剂),确定并控制潜在的质量风险,消除或者不断降低中间产品给药品质量造成的风险,公司就液体制剂车间生产过程进行了风险评估、风险控制和管理。 2.目的 对影响生产过程控制的因素进行评价,对可能的危害进行判定,对于每种危害可能产生损害的严重程度、危害的发生概率和可检测性进行估计,在某一风险水平不可接受时,建议采取降低风险的措施,在日常管理中进行控制。 3.风险管理人员及其职责分工 4.风险分析 . .主要从人流与物流、公用工程、设备、清洁消毒、物料系统、空调系统共六个方面进行分析,详见下图: 风险等级判定: 采用RPN(严重程度、发生频率和可发现的可能性等级三者乘积)进行风险优先数量等级判定。 危害性(S):根据对药品质量的影响判定→ 1~5分 对产品质量产生严重影响,使药品不合格,5分 造成产品不合格,需对产品进行返工,4分 造成产品收率达不到要求,3分 对产品产生轻微影响,2分 对产品质量几乎无影响,1分 发生的可能性(P):根据出现频次判定1~5分 每周出现1次5分 每月出现1次4分 每季度出现1次3分 每半年出现1次2分 每年出现1次1分 可发现性(D):根据风险发生时能够检测的程度判定1~5分 难以检测5分 需要专职人员检测4分 操作员工能够检测3分 一般人员容易检测2分 有仪器仪表检测1分 风险等级判断 1-8分为低等级风险,可以接受;9-36分为中等级风险,由车间工艺员和现场QA加大复核检查的频次予以控制;37-80分为高等级风险,予以特别关注,计划制订专门的管理制度、操作规程以及采用预防和纠正措施予以控制,并通过对风险进行控制后的风险进行两次风险评估,确认其RPN分数已下降到可以接受的程度。 风险评级及措施要求 固体制剂车间多品种共线生产风险评估报告 编号:FX-SC-2015-002 固体制剂车间多品种共线生产 风险评估报告 起草部门及职务责任人签名起草日期 审核部门及职务责任人签名审核日期批准人责任人签名批准日期 吉林九鑫制药股份有限公司 二零一五年五月 固体制剂车间多品种共线生产风险评估报告 目录 1 概述 1.1相关品种明细 1.2具体的设备明细 2 风险评估标准 3 找出评估风险点 4 对提出的风险点进行评估 4.1原料性质风险评估 4.2设备清洁风险评估 5 结论 1 概述 公司生产的口服固体制剂品种有39个品种, 39个批准文号,其中片剂品种2个,胶囊剂品种3个,颗粒剂品种2个,水丸、水蜜丸、浓缩丸剂品种5个,大蜜丸剂品种27个。常年生产的品种4个。公司配备了先进的符合2010版GMP 要求的生产设备,配套了完善的GMP 文件软件系统,对生产线的各个关键操作环节进行同步监控。 1.1相关品种明细如下: 剂型 品种名称 丸剂 大蜜丸 偏瘫复原丸、补中益气丸、大山楂丸、二十七味定坤丸(定坤丸)、附子理中丸、舒肝丸、太极丸、天王补心丸、小活络 丸、杞菊地黄丸、大活络丸、回天再造丸、琥珀安神丸、八珍益母丸、桂附地 黄丸、参桂再造丸、通宣理肺丸、艾附暖宫丸、橘红丸、牛黄上清丸、利膈丸、 紫蔻丸、朱砂安神丸、百合固金丸、十 全大补丸、人参归脾丸、六味地黄丸、 水丸、 水蜜丸、浓 缩丸 清热暗疮丸、六味地黄丸、更年宁、人 参再造丸、银翘解毒丸 胶囊剂 胃乐新胶囊、乙肝扶正胶囊、速感宁胶 囊 片剂 咽炎片、消积健儿片 颗粒剂 益肾灵、通脉颗粒 1.2具体的设备明细如下: 序号 设备名称 型号规格 设备编号 生产厂家 1 高效强力性竖式混合机 SGTJ-046 黑龙江省迪尔 制药机械有限公司 2 不锈钢粉碎机 FS-320 SGTJ-013 丹东市制药机械有限公司 深圳市XXX印刷有限公司 纸质圣诞印刷产品 质量风险评估报告 报告起草:年月日报告审核:年月日报告批准:年月日 深圳市XXX印刷有限公司 二0一六年八月 目录 1.纸质圣诞印刷产品相关信息 (1) 2.纸质圣诞印刷产品质量风险概述 (1) 3.纸质圣诞印刷产品质量风险识别 (1) 4.风险分析 (3) 4.1纸质圣诞印刷产品风险失败模式建立 (3) 4.2纸质圣诞印刷产品风险分析 (4) 4.2.1人员风险 (4) 4.2.2设备、仪器风险 (5) 4.2.3纸质圣诞印刷产品用物料风险 (6) 4.2.4纸质圣诞印刷产品生产操作方法风险 (7) 4.2.5纸质圣诞印刷产品生产环境风险 (10) 4.2.6纸质圣诞印刷产品检验(测量)风险 (13) 5.评估总结论与建议 (14) 6.本风险评估依据与资料收集范围 (15) 产品名称:纸质圣诞印刷产品产品阶段:生产全过程评估小组成员: 评估日期: 1.纸质圣诞印刷产品相关信息 纸质圣诞产品基本信息:通用名称:圣诞装饰品,本产品主要用于圣诞节各种圣诞树及圣诞礼物的挂件装饰用。 我公司生产的纸质圣诞印刷产品的工艺规程、质量标准以及包装标签上的信息均符合国家及相关客户标准。 2.纸质圣诞印刷产品质量风险概述 本报告纸质圣诞印刷产品质量风险进行系统的分析评估,对纸质圣诞印刷产品涉及的生产过程所有可能出现的风险进行评估,确定重点控制的目标,制定纠正和预防措施,对于高风险和中等风险的没有管理措施的必须确定降低风险的措施,低风险加强生产过程控制,确保产品质量,降低风险发生的可能性,提高可识别性,将风险控制在可接受水平。如果采取风险控制措施和预防措施后风险仍不可接受,应重新制定降低风险的措施和办法。本风险评估资料来源于公司纸质圣诞印刷产品质量档案,历年生产记录,各种涉及的偏差、变更、验证、工艺规程、质量标准涉及等纸质圣诞印刷产品生产的全部记录资料。 3、纸质圣诞印刷产品风险识别 纸质圣诞印刷产品质量风险的识别用鱼骨图来描述,根据鱼骨图(见下图)逐一展开分析和评价,找出风险点进行控制,把降低风险的措施落实到每个环节。 目录 1 前言 (1) 2 总则 (3) 2.1编制原则 (3) 2.2编制依据 (3) 2.3环境风险评估的程序 (6) 3 资料准备与环境风险识别 (7) 3.1企业基本信息 (7) 3.2企业周边环境风险受体情况 (11) 3.3涉及环境风险物质情况 (13) 3.4生产工艺 (32) 3.5安全生产管理 (41) 3.6现有环境风险防控与应急措施情况 (41) 3.7现有应急物资与装备、救援队伍情况 (45) 4 突发环境事件及其后果分析 (49) 4.1突发环境事件情景分析 (49) 4.2突发环境事件情景源强分析 (57) 4.3释放环境风险物质的扩散途径、涉及环境风险防控与应急措施、应急 资源情况分析 (62) 4.4突发环境事件危害后果分析 (69) 5.现有环境风险防控和应急措施差距分析 (72) 5.1环境风险管理制度 (72) 5.2环境风险防控与应急措施 (75) 5.3环境应急资源 (77) 5.4历史经验教训总结 (80) 5.5需要整改的短期、中期和长期项目内容 (81) 6.完善环境风险防控和应急措施的实施计划 (84) 7.企业突发环境事件风险等级 (88) 7.1环境风险物质数量与临界量比值值 (88) 7.2生产工艺与环境风险控制水平 (92) 7.3环境风险受体敏感性性(E) (97) 7.4企业环境风险等级划分 (99) 1 前言 ................制药股份有限公司的前身——河南...制药厂始建于1969 年,1997 年股改成立股份公司,现已发展成为国家级制药骨干企业之一,...市高新技术产业。公司位于...县城东南部。主营业务为中西药品和化学原料药的研发、生产和销售。主要产品为大容量注射液、小容量注射液、片剂、胶囊剂、颗粒剂、露剂、酊剂和原料药等八大剂型160多个品种,全部生产车间均多次通过国家GMP认证。主导产品有国家级新药援生力维片、精制冠心颗粒、清热解毒颗粒、马来酸曲美布汀原料,国家中药保护品种有猫爪草胶囊、伤痛宁、桑姜感冒片等。产品销售全国二十多个省区。其中马来酸曲美布汀片(商品名:援生力维)是公司与上海医工院联合研制的国家级新一代胃肠功能调节剂,自上市以来产品销量稳居市场前列,自2004年以来连续二次被评为河南省名牌产品、...市高新技术产品;在此基础上,公司对此产品进行了深度开发,2010年5月公司联合中国药科大学开发的具速释部分的马来酸曲美布汀缓释片及其制备工艺获得国家发明专利,已获得新药证书并已经上市。马来酸曲美布汀原料质量产量国内领先,通过韩国、日本、墨西哥官方GMP认证。出口亚洲、欧洲、美洲多个国家,销量居国内前列。 ................制药股份有限公司1997年进行股份制改造,2001年与上海开开实业公司增资扩股组建为县公司,目前属大型二类企业,注册资本8400万元,总资产为1.6亿元,年销售收入为2亿多元, 质量风险评估 编号: 软袋车间产品生产过程质量风险评估 日年月起草人: 年日月审核人: 批准人:年日月 目录 1 风险评估的目的 2风险评估小组组成及职责3风险分级情况 3.1损害的严重度的分类3.2 危害发生概率的分类3.3风险评价准则: 4 风险评估内容 4.1物料管理的风险评估4.2生产管理风险评估 4.3空调系统和工艺用水系统的风险评估 质量管理风险评估4.4 1 风险评估的目的 通过对软袋车间整个生产过程进行风险评估,对可能存在的风险和隐患进行分析、评价并采取相应的措施,使风险消除或降低至安全等级,达到可控制的水平,以减少质量风险的发生概率。 2 风险评估小组的组成及职责: 姓组内职部门、职岗职 负责组织风险评估方案的起草、实施并制定风险等级评估以及风险预防措施的方案,负责组织风险评估报组质量管理部经 的汇总及给出风险评估报告的总评价,批准风险评估告 组储运部主共同从物料进厂验收、入库、贮存到发送全过程等方提出风险评估内容,对其进行风险分析,风险分级及QA质量管理组险评价。通过对风险的评价,制定降低风险、控制风储运部经组 的措施 制造部副经组从空调系统、工艺用水系统方面提出风险评估内容, 其进行风险分析,风险分级及风险评价。通过对风险评价,制定降低风险、控制风险的措施 组30车间主共同从生产管理中的物料(从仓库至车间的运输、物在车间的贮存传递使用全过程人员操作生产QA 组质量管理程中物料交叉污染和混淆、生产用仪器或设备、各生工序等方面的风险评估内容,对其进行风险分析,风分级及风险评价。通过对风险的评价,制定降低风险控制风险的措施 组质量管理QA从质量管理文件的角度提出质量管理方面的风险评估容,对其进行风险分析,风险分级及风险评价。通过对风险的评价,制定降低风险、控制风险的措施。. 原料药工艺验证通则 在原料药生产过程中,工艺验证是提供文件化的证据,证明用于原料药生产的人员、材料、设备、方法、环境条件以及其他有关公用设施的组合,可以持续生产出符合预定用途和注册要求的产品。本通则对ICH-Q7A中关于工艺验证的方法及工艺验证的执行进行了详细解析,包括工艺验证目的,针对不同情况的工艺验证方法,以及使用适当的风险管理工具进行工艺风险评估和相应工艺验证方案的实施。 1 工艺验证的目的 工艺验证是利用文件化的证据,证明将生产无菌原料药产品的人员、材料、设备、方法、环境条件以及其他有关公用设施进行有机组合,可以持续地生产出符合预定用途和注册要求的产品,工艺稳定可靠,符合GMP要求。 工艺验证方案将对工艺验证过程中的中间控制及检查结果进行合理设计并进行详细记录。通过工艺验证可以建立定期的生产工艺再评估工作,并对每一步的生产工艺进行监控,以确保产品始终符合既定的标准和质量特性。同时,对中间体进行检测和成品检测,使工艺过程始终处在良好的控制之下。 2 工艺验证的分类 工艺验证有三种类型,分为前验证、同步验证、回顾性验证。其中,前验证是首选的验证方法。 2.1 前验证 对于ICH-Q7A定义的所有原料药工艺,一般都采用前验证。对一种原料药实施的工艺验证应该在商业销售之前完成。 2.2 同步验证 特殊情况下,可以采用同步验证的方式进行产品的工艺验证,例如,所验证原料 药产品生产批次较少或不经常生产,或是因为经过验证的生产工艺变更引起的工艺验证,可以采用同步验证方式。在同步验证完成之前,如果有周密的监控和检验,原料药可以放行并用于制剂的生产,并且可以进行商业销售。 2.3 回顾性验证 回顾性验证仅适用于已生产多年、工艺足够成熟的产品,产品的关键工艺参数和控制点也已完全确定,并且在产品生产过程中,相关的工艺、设备、原料、厂房设施均没有发生重大的变化。 回顾性验证的验证方案要涵盖可接受标准和详细的生产信息。对于不合格的批号和存在异常趋势的批号要进行调查。回顾性验证批数的选择要符合统计学,一般规律是要求20~30批,但公司验证的批数可以不是20~30批,只要有统计数据或其他依据支持此验证批数的选择方式即可。 3 工艺过程风险评估 风险评估将所识别和分析的风险与给定的风险标准进行对比,确定生产产品的质量。 3.1 危害分析 组建专门的风险评估团队,开展二个阶段的危害分析: 在第一阶段中,评估团队将对无菌原料药生产过程中所涉及的物料、活动、设备、储存、分发和预计用途进行分析。将写出每一步骤可能引入、增加或控制的潜在的危害(生物、化学和物理)的清单。 在危害分析中,可能包括但不限于下述问题:(1)可能发生的危害及其危害的严重性;(2)危害存在的定性和/或定量评估;(3)相关微生物的残存或繁殖;(4)毒性、物理或化学试剂的生产或在药品中的残留;(5)上述内容的引导条件。 第二阶段,危害评估的执行,即潜在危害的严重性和发生的可能性的评估。 评估团队将决定在关键工艺控制点计划中说明存在有哪些潜在的危害,采取什么控制措施。控制一个具体的危害可能要求至少有一个控制措施,一个具体的控制措施 目的 确认供应商审计的围及程度,识别供应商及物料采购选择质量风险,对风险进行分级,根据等级大小,进行分析、评估,确保关键风险要素能够得到有效控制,以降低供应商带来的质量风险,并为及时更换供应商提供依据。 围 公司生产品种所涉及的原辅料、包材的供应商均在此风险评估的围,重点是评估供应商的质量管理体系和所采购物料的风险等级。 责任 质量管理部、供销部 容 1供应商风险评估:包括质量保证能力评估、供货历史评估、维护性评估。 2采购物料风险评估:分为关键性物料、影响产品质量物料、不影响产品质量物料等三级质量风险评估。 3风险评估小组组员及职责 4风险评估程序 风 险 管 理 工 具 启动质量风险管理 5 供应商质量风险评估项目、风险分析原则及标准: 一、项目确定原则: 1.供应商系统设计性能检测项目 2.生产工艺设计储存条件对系统的要求 3.《洁净厂房设计规》GB50073-2001 4. 《药品生产质量管理规》2010版 二、评估标准: 根据我公司生产所用的供应商,对供应商相关资质、机构与人员、厂房和设施设备、物料管理、生产管理、质量管理、运输与交货七个重点项目用帕累托图分析法进行分析。分析供应商所存在的问题,分为3类,A类属于关键问题,累计分数在0~80%;B类问题属于次要问题,累计分数在80%~90%;C类问题属于一般问题,累计分数在90%~100%。 年月日至年月日,风险评估小组人员对供应商系统按照重点项目进行风险评估,各关键要素的风险分析,评估及结果见下表: 评分标准 0分-------- 未有文件 ; 1分 -------- 手写的程序或文件(未受控) ; 2 分-------- 不足夠,需要改善 ; 3 分-------- 备注,需要关注; 4分-------------- 满意; N/A ------------- 不适用. 风险评估表生产工艺质量风险评估
工艺风险评估与验证案例
产品质量风险评估报告
片剂工艺验证方案
关于质量风险评估报告
某原料药工艺风险评估
药品生产过程质量风险评估报告模板
液体制剂生产过程质量风险评估报告
固体制剂车间多品种共线生产风险评估报告
印刷质量风险评估报告
化学合成制药厂风险评估分析
质量风险评估
原料药工艺验证
供应商风险评估方案报告
相关主题
文本预览