
荧光淬灭法测定Ni(II)
摘要:
目的运用荧光淬灭法检测工业废水①中的Ni(II)是否达标。
方法镍对邻二氮菲的荧光具有淬灭的特性,本文采取标准曲线法,首先测定一系列邻二氮菲的标准溶液的荧光强度,得到以荧光强度(F)与邻二氮菲浓度(c)的曲线,再测定用过量的邻二氮菲处理的Ni(II)试样的荧光强度,最后间接计算出Ni(II)试样的浓度。
通过标准曲线法课以有效地扣除背景,减小误差,结果相对准确,标准曲线的r2约为0.99925②,通过与环境监测部门给出的数据对比,相对误差较小,结果值得信赖。
关键词:镍邻二氮菲荧光淬灭法
Abstract:
Purpose use the fluorescence quenching method to detect Ni (II) in the natural lake.
Method this method is based on the quenching of fluorescence of phenanthroline due to the formation of complex Ni (II)-phen. Firstly, determine the fluorescence intensity of a series of o-phenanthroline standard solution, so we obtain a standard curve about fluorescence intensity(F) and o-phenanthroline concentration (c) . Then determine the fluorescence intensity of Ni (II) sample with excess o-phenanthroline treatment, and finally indirectly calculate the Ni (II) sample concentration.
Through the standard curve method we can effectively deduct the background, reduce errors, the result is relatively accurate, the standard curve r2 is about 0.99925, and by the comparison of the data given by the environmental monitoring department, the relative error is small, the results reliable.
Key words: nickel phenanthroline fluorescence quenching
引言:镍是人体必需的微量元素之一, 对体内某些酶有激活作用及刺激造血机能和促进红细胞再生的作用。但过量亦会中毒③。环境中镍的主要污染来源为:镍矿的开采和冶炼;合金钢的生产和加工过程;煤、石油燃烧时排放烟尘中;电镀、镀镍的生产过程④。测定镍的方法有原子吸收法和ICP-AES法、丁二酮肟分光光度法、极谱催化波等⑤。本文基于镍对邻菲罗啉自身荧光淬灭的作用,进行了痕量镍的测定。该方法具有操作简便,测定快速,重现性好,灵敏度高等特点。应用该方法测定了工业废水中的痕量镍,得到了满意的结果。
①以某冶金厂的废水为测定样本;
②本文中的测定数据均属由在实验中的分子荧光法测定奎宁含量的实际数据类比之后虚拟而得;
③孙成均. 生物材料检验[M] . 北京: 人民卫生出版社, 2006. 49;
④镍有何毒性与危害?南宁环保局网络课堂2005-06-10;
⑤《PAN- Rh6G 能量转移荧光猝灭法测定痕量镍》,宁玲, 吕昌银, 陈云生, 范翔, 贺元文,2008.05
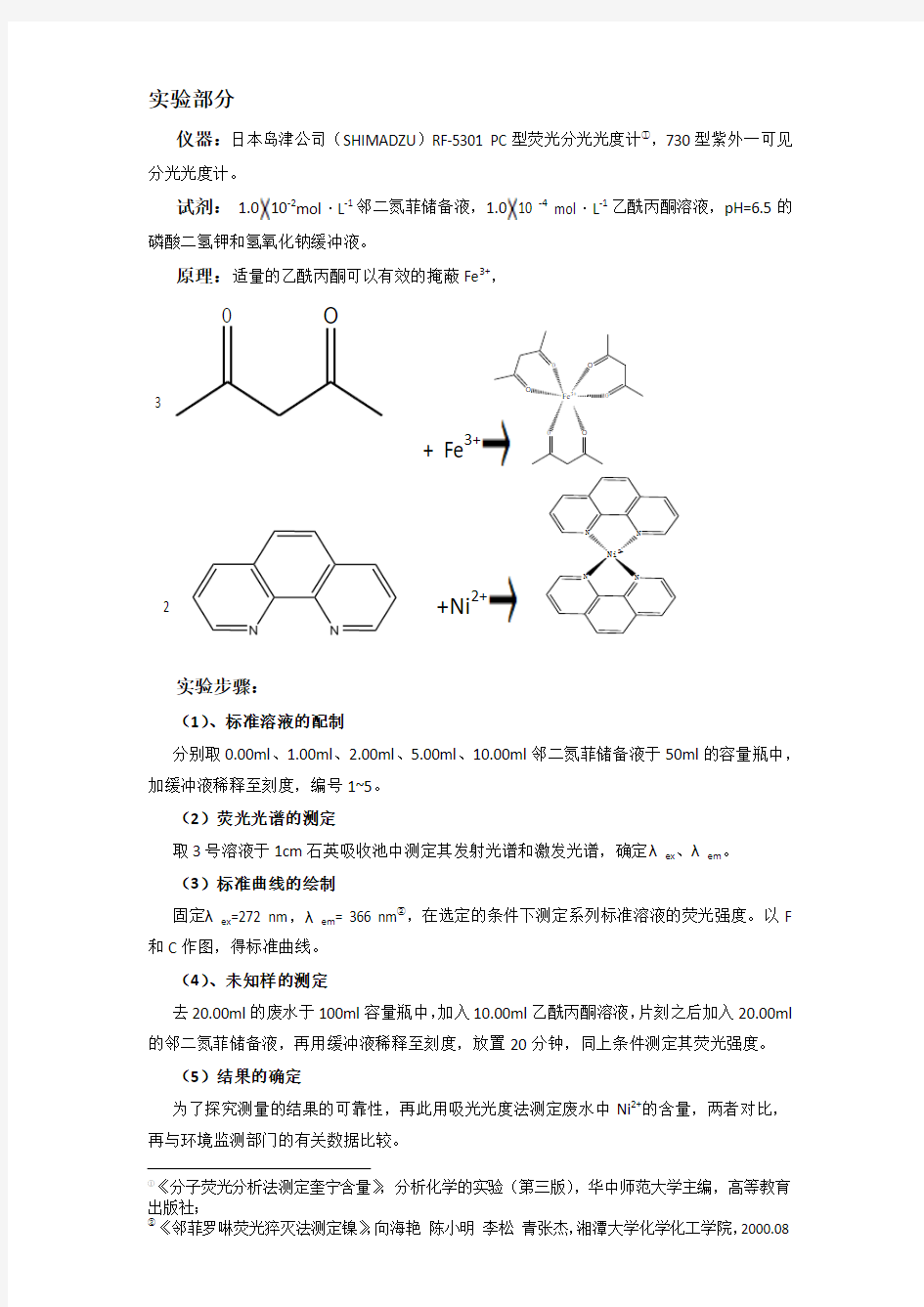
仪器:日本岛津公司(SHIMADZU )RF-5301 PC 型荧光分光光度计①,730型紫外一可见
分光光度计。
试剂: 1.010-2mol ·L -1邻二氮菲储备液,1.010 -4 mol ·L -1乙酰丙酮溶液,pH=6.5的
磷酸二氢钾和氢氧化钠缓冲液。
原理:适量的乙酰丙酮可以有效的掩蔽Fe 3+,
O
O
O
O O
O
O
O
Fe 3+
N
N
Ni 2
+
实验步骤:
(1)、标准溶液的配制
分别取0.00ml 、 1.00ml 、2.00ml 、5.00ml 、10.00ml 邻二氮菲储备液于50ml 的容量瓶中,加缓冲液稀释至刻度,编号1~5。
(2)荧光光谱的测定
取3号溶液于1cm 石英吸收池中测定其发射光谱和激发光谱,确定λ
ex 、λem 。
(3)标准曲线的绘制
固定λ
ex =272 nm ,λem = 366 nm
②
,在选定的条件下测定系列标准溶液的荧光强度。以F
和C 作图,得标准曲线。
(4)、未知样的测定
去20.00ml 的废水于100ml 容量瓶中,加入10.00ml 乙酰丙酮溶液,片刻之后加入20.00ml 的邻二氮菲储备液,再用缓冲液稀释至刻度,放置20分钟,同上条件测定其荧光强度。
(5)结果的确定
为了探究测量的结果的可靠性,再此用吸光光度法测定废水中Ni 2+的含量,两者对比,再与环境监测部门的有关数据比较。
①
《分子荧光分析法测定奎宁含量》,分析化学的实验(第三版),华中师范大学主编,高等教育出版社; ②
《邻菲罗啉荧光猝灭法测定镍》,向海艳 陈小明 李松 青张杰,湘潭大学化学化工学院,2000.08
+ Fe 3+
3
2 +Ni 2+
激发光谱和发射光谱本处略去。
绘制标准曲线如下:
F
C 根据测定的试样中邻二氮菲的浓度,和添加到试样中实际邻二氮菲的量,计算出淬灭部分的邻二氮菲,在间接求出Ni2+的浓度,与分光光度法测定的含量相比较。
结果分析:
通过荧光淬灭法间接测定Ni2+的浓度,其准确性取决于其与邻二氮菲的络合平衡常数,干扰离子与邻二氮菲的络合,温度,pH值。通过稳定室温在20℃左右,加入乙酰丙酮做掩蔽剂,和添加缓冲液,有利于消减误差,使得结果更可靠。
实验中合适的条件并不是已知的,而是通过多次试验的数据记录,处理而得,本文中借鉴一些专家给出的条件,得以是的测定工作量减小了许多。
事实证明,此法的测定结果与分光光度法测定的数据相对误差较小,与环境监测部门公布的数据也比较接近。
分子荧光光谱法实验报告 一、实验目的 1.掌握荧光光度计的基本原理及使用。 2.了解荧光分光光度计的构造和各组成部分的作用。 3.掌握分子荧光光度计分析物质的特征荧光光谱:激发光谱、发射光谱的测定方法。 4.了解影响荧光产生的几个主要因素。 5.学会运用分子荧光光谱法对物质进行定性和定量分析。 二、实验原理 原子外层电子吸收光子后,由基态跃迁到激发态,再回到较低能级或者基态时,发射出一定波长的辐射,称为原子荧光。对于分子的能级激发态称为分子荧光,平时所说的荧光指分子荧光。 具有不饱和基团的基态分子经光照射后,价电子跃迁产生荧光,是当电子从第一激发单重态S1的最低振动能级回到基态S0各振动能级所产生的光辐射。 (1)激发光谱 是指发光的某一谱线或谱带的强度随激发光波长(或频率)变化的曲线。横坐标为激发光波长,纵坐标为发光相对强度。 激发光谱反映不同波长的光激发材料产生发光的效果。即表示发光的某一谱线或谱带可以被什么波长的光激发、激发的本领是高还是低;也表示用不同波长的光激发材料时,使材料发出某一波长光的效
率。荧光为光致发光,合适的激发光波长需根据激发光谱确定——激发光谱是在固定荧光波长下,测量荧光体的荧光强度随激发波长变化的光谱。获得方法:先把第二单色器的波长固定,使测定的λem不变,改变第一单色器波长,让不同波长的光照在荧光物质上,测定它的荧光强度,以I为纵坐标,λex为横坐标所得图谱即荧光物质的激发光谱,从曲线上找出λex,,实际上选波长较长的高波长峰。 (2)发射光谱 是指发光的能量按波长或频率的分布。通常实验测量的是发光的相对能量。发射光谱中,横坐标为波长,纵坐标为发光相对强度。 发射光谱常分为带谱和线谱,有时也会出现既有带谱、又有线谱的情况。发射光谱的获得方法:先把第一单色器的波长固定,使激发的λex不变,改变第二单色器波长,让不同波长的光扫描,测定它的发光强度,以I为纵坐标,λem为横坐标得图谱即荧光物质的发射光谱;从曲线上找出最大的λem。 (3)荧光强度与荧光物质浓度的关系 用强度为I0的入射光,照射到液池内的荧光物质时,产生荧光,荧光强度If用仪器测得,在荧光浓度很稀(A 三、实验试剂和仪器试剂:罗丹明B乙醇溶液;1-萘酚乙醇溶液;3,3’-Diethyloxadicarbocyanine iodide:标准溶液,10μg/ml, 20μg/ml,30μg/ml,40μg/ml和未知浓度;蒸馏水;乙 醇。 仪器:Fluoromax-4荧光分光光度计;1cm比色皿;
分子荧光法测定水杨酸和乙酰水杨酸 一、实验目的 1、学习荧光分析法的基本原理。 2、掌握应用分子荧光法测定乙酰水杨酸、水杨酸的分析方法。 3、了解F-3501型荧光分光光度计的主要结构及工作原理,掌握其正确的使用方法。 二、实验原理 某些物质经紫外光或波长较短的可见光照射后,会发射出较入射波长更长的荧光。荧光光谱反映了物质的特性,建立在测量荧光光谱基础上的分析方法称为荧光分析法。当进行荧光测定时,总要选择不同波长的光波进行测定,即一个为激发光——物质所吸收的光;另一个为物质吸收后发出的光称为发射光或荧光。 对同一物质而言,在稀溶液(即A = abc < 0.05)中,荧光的强度F与该物质的浓度C 有以下关系: F = 2.3φabcI0 式中φ为荧光过程的量子效率,a为荧光分子的吸光系数,b为试样的吸收光程,I0为入射光的强度。当I0及b 不变时,上式变为: F= KC 其中,K为常数。 荧光分析法具有灵敏度高(一般超过分光光度法2~3个数量级)、取样少、方法快速等特点,现已成为食品、生物医药、天然产品、农业、环境保护、化工等领域中的重要分析方法之一。但由于许多物质本身不会发生荧光,故在使用范围上受到一定的限制。 乙酰水杨酸(ASA,即阿司匹林)水解能生成水杨酸(SA),而在乙酰水杨酸中,或多或少都存在着水杨酸。由于两者都有苯环,也有一定的荧光效率,因而在以三氯甲烷为溶剂的条件下,可用荧光法进行测定。从乙酰水杨酸和水杨酸的激发光谱和荧光光谱中可以发现:乙酰水杨酸和水杨酸的激发波长和发射波长均不同,利用此性质,可在各自的激发波长和发射波长下分别测定。 三、仪器与试剂 1、仪器 F-3501型荧光分光光度计、电子天平、离心机、真空泵、石英比色皿、移液管、棕色容量瓶、比色管、烧杯、砂芯抽滤装置、量筒、洗耳球、洗瓶等。
农药残留分析 农药残留分析复习题 简答题 1、写出各种硅胶代号及其意义 a.硅胶H—不含粘合剂 b.硅胶G—硅胶和锻石膏混合而成 c.硅胶HF254—不含粘合剂,含荧光 指示剂,254nm呈现强绿色荧光背景 d.硅胶GF254—同时含粘合剂和荧光指示剂,254nm光致发光,呈强的绿色荧光背景 e.硅胶HF254+336—不含粘合剂,含无机荧光剂,在254nm和336nm呈现荧光背景 2、简述农药残留分析常用的气相色谱仪检测器的类型及其特点 (1)火焰离子化检测器(FID)特点:质量型,准通用型;最高操作温度(℃)450最低检测限丙烷<5 pg 碳/s;线性范围107(±10%)主要用途各种有机化合物的分析,对碳氢化合物的灵敏度高 (2)热导检测器(TCD)特点:浓度型,通用型;最高操作温度(℃) 400;最低检测限<400 pg/ml,壬烷:20000 mv?ml/mg;线性范围丙烷:104(±5%);主要用途适用于各种无机气体和有机物的分析,多用于永久气体的分析。 (3)电子俘获检测器(ECD)特点:浓度型,选择型;最高操作温度(℃) 400;低检测限六氯苯<0.04 pg/ml;范围>104;主要用途适合分析含电负性元素或基团的有机化合物,多用于分析含卤素化合物。 (4)氮磷检测器(NPD)特点:质量型,选择型;操作温度(℃) 400;低检测限用用偶氮苯和马拉硫磷的 混合物测定:<0.4 pg氮/s; <0.2 pg磷/s;范围>105;要用途适合于含氮和含磷化合物的分析。 (5)火焰光度检测器(FPD)特点:质量型,选择型;高操作温度(℃) 250;低检测限用十二烷硫醇和 三丁基膦酸酯混合物测定:<20 pg硫/s,<0.9 pg磷/s;性范围硫:>105磷:>106;要用途适合于含硫、含磷和含氮化合物的分析。 3、简述酶抑制法测定农药残留的基本原理 乙酰胆碱酯酶与胆碱能神经的生理功能极为密切。其生理功能是:当神经冲动到达胆碱能神经末梢时,突触小泡内的Ach(乙酰胆碱)外排至突触间隙,作用于突触后膜的胆碱能受体,引起下一级神经元或效应器的激发。在正常生理条件下,Ach完成传递冲动作用后,随即被突触后膜上的AchE在数毫秒内水解,生成乙酸和胆碱,水解产物与加入的试剂反应产生颜色。反之,式样提取剂中含有一定量的有机磷或氨基甲酸酯类农药,酶的活性就被抑制,式样中加入的机制就不能被水解,从而不显色或颜色很浅,用分光光度计测定吸光度,计算出抑制率,就可判断农药残留情况。 4、简述超声波萃取原理 超声波辅助萃取:是利用超声波辐射压强产生的强烈空化效应、机械振动、扰动效应、高的加速度、乳化、扩散、击碎和搅拌作用等多级效应,增大物质分子运动频率和速度,增加溶剂穿透力,从而加速目标成分进入溶剂,促进提取的进行。 名词解释 1、农药残留:指由于农药的应用而残存于生物体、农产品和环境中的农药亲体及其具有毒理学意义的杂 质、代谢转化产物和反应物等所有衍生物的总称。 农药残留毒性:因摄入或长时间重复暴露农药残留而对人、畜以及有益生物产生急性或慢性中毒 2、农药纯品:能够真正反映化合物特性的高纯度物质。 农药标准物质:具有一种或多种足够均匀和很好确定了的特性值,用以校准设备,评价测量方法或给材料赋值的材料或物质。 3、最大残留限制(MRL) :指由食品营养标准委员会推荐的,食品或动物饲料中允许的农药残留物的最大 浓度(毫克/千克)。是根据在毒理学上认为可以接受的食品农药残留量制定的。 每日允许摄入量(ADI) :指在一生中,对消费者健康没有可感知危险的日摄入量。 论述题 1、论述农药标准品常用的制备方法p62 ①重结晶法(适于除去农药原粉中的少量杂质) ②蒸馏法 ③柱层析法(其特点:应用范围广、操作简便、分离效率高) ④区域熔融法 ⑤升华法(此法在农药纯品制备中应用较少) ⑥薄层色谱法(常用来制备毫克级农药纯品) 2、写出样品提取技术的类型及其提取剂的要求 (1)溶剂提取法包括液液提取和固液提取 固液提取常用以下方法:①索式提取法②振荡提取法③组织捣碎法④消化提取法 (2)固相提取法
2341 农药残留量测定法 第五法药材及饮片(植物类)中禁用农药多残留测定法 1. 气相色谱-串联质谱法 色谱条件用(50%苯基)-甲基聚硅氧烷为固定液的弹性石英毛细管柱(柱长为30m,柱内径为0.25mm,膜厚度为0.25μm)。进样口温度250℃,不分流进样。载气为高纯氦气(He)。进样口为恒压模式,柱前压力为146kPa。程序升温:初始温度60℃,保持1分钟,以每分钟10℃的速率升温至160℃,再以每分钟2℃ ) , % 监测离子对、碰撞电压(CE)见附表2。为提高检测灵敏度,可根据保留时间分段监测各农药。 3. 对照溶液的制备 3.1 混合对照品溶液的制备精密量取禁用农药混合对照品溶液(已标示各相关农药品种的浓度)1ml,置20ml量瓶中,用乙腈稀释至刻度,摇匀,即得。
3.2气相色谱-串联质谱法分析用内标溶液的制备取磷酸三苯酯对照品适量,精密称定,加乙腈溶解并制成每1ml含1.0mg的溶液,即得。精密量取适量,加乙腈制成每1ml含0.1μg的溶液。 3.3 空白基质溶液的制备取空白基质样品,同供试品溶液的制备方法处理制成空白基质溶液。 3.4 基质混合对照溶液的制备分别精密量取空白基质溶液1.0ml(6份),置氮吹仪上,40℃水浴浓缩至约0.6ml,分别加入混合对照品溶液10μl、20μl、50μl、100μl、150μl、200μl,加乙腈稀释至l ml,涡旋混匀,即得。 4. 供试品溶液的制备 4.1 直接提取法 取供试品粉末(过三号筛)5g,精密称定,加氯化钠1g,立即摇散,再加入乙腈50ml,匀浆处理2分钟(转速不低于每分钟12000转),离心(每分钟4000转),分取上清液,沉淀再加乙腈50ml,匀浆处理1分钟,离心,合并两次提取的上清液,减压浓缩至约3~5ml,放冷,用乙腈稀释至10.0ml,摇匀,即得。 4.2 快速样品处理法(QuEChERS)法 取供试品粉末(过三号筛)3g,精密称定,置50ml聚苯乙烯具塞离心管中,加入1%冰醋酸溶液15ml,涡旋使药粉充分浸润,放置30分钟,精密加入乙腈15ml,涡旋使混匀,置振荡器上剧烈振荡(每分钟500次)5分钟,加入无水硫酸镁与无水乙酸钠的混合粉末(4:1)7.5g,立即摇散,再置振荡器上剧烈振荡(每分钟500次)3分钟,于冰浴中冷却10分钟,离心(每分钟4000转)5分钟,取上清液9ml,置预先装有净化材料的分散固相萃取净化管[无水硫酸镁900mg,N-丙基乙二胺300mg,十八烷基硅烷键合硅胶300mg,硅胶300mg,石墨化碳黑90mg]中,涡旋使充分混匀,置振荡器上剧烈振荡(每分钟500次)5分钟使净化完全,离心(每分钟4000转)5分钟,精密吸取上清液5ml,置氮吹仪上于40℃水浴浓缩至约0.4ml,加乙腈稀释至1.0ml,涡旋混匀,滤过,取续滤液,即得。 4.3固相萃取法 固相萃取净化方式包括以下三种: 方式一:量取直接提取法制备的供试品溶液3~5ml,置于装有分散型净化材料的净化管[无水硫酸镁1200mg,N-丙基乙二胺300mg,十八烷基硅烷键合硅胶
1我报告的题目是基于碌酸化多肽降解受阻的石墨稀/多肽复合焚光探针用于蛋白激酶活性及抑制作用分析 2蛋白激酶催化作用下的蛋白质憐酸化是生物体内翻译后修饰的重要方式之一。蛋白质的憐酸化和去磷酸化这一可逆过程几调节着细胞的发育、增殖、分化、信号转导、神经活动、肌肉收缩、细胞凋亡及肿瘤发生等过程在内的大部分生命活动。非正常的磷酸化会导致人体产生各种疾病,例如癌症或老年痴呆症等。因此,准确灵敏地检测过度磷酸化、分析蛋白激酶活性和高通量蹄选高效抑制剂对于人类一些重大疾病的早期诊断和治疗极为重要。 传统用于蛋白激酶活性分析的方法依赖于放射性元素标记伽马-32P-ATP法,由于放射性物质对环境及其人体的危害而随之被取代,基于憐酸特异性识别抗体的免疫技术[146]、突光分析方法表面等离子共振技术[184,185]以及质谱等都能有效用于蛋白激酶的活性分析。 石墨稀(Graphene),是从石墨材料中剥离出来的由碳原子组成的二维晶体, 是目前己知世界上强度最高的材料。石墨稀具有独特的电子、机械、热力学特性 在化学、质量、压力等传感应用中独具优势,。与碳纳米管相似,石墨烯对有机 荧光分子表现了有效的突光淬灭效果,这一过程中同时发生了激发态突光分子与 石墨烯表面之间的能量转移和电子转移。2010年诺贝尔物理学奖 氧化石墨烯薄片是石墨粉末经化学氧化及剥离后的产物,氧化石墨烯是单一的原子层,仍然具有淬灭荧光的效果 3本文首次提出了一种基于多肽/石墨烯突光浮灭机制和接肽酶降解作用的激 酶活性分析方法。酪蛋白激酶CKII是一种重要的丝氨酸/苏氨酸选择性蛋白激酶, 能磷酸化160种不同的蛋白质,我们以CKII为蛋白激酶模型。FITC 标记的CKII底物多 肽,FITC-peptide(FITC-RRRADDSDDDDD),能与GO发生有效的突光浮灭。幾肽酶CPY是一类 肽链端解酶,作用于任何一个C-末端残基,从肽链的C端开始逐个降解,释放出 游离氨基酸。FITC-peptide在CPY的消化作用下释放出游离的FITC分子,在 高离子强度环境下,游离FITC分子与GO之间的吸附较小而保持相当强的焚光。 而当FITC-peptide在CKII/ATP催化发生磷酸化,有效地阻碍CPY在憐酸化丝氨 酸位点的降解,使得FITC-多肽更容易与GO结合导致焚光淬灭。 1羧肽酶CPY 作用于任何一个C-末端残基逐个降解释放游离氨基酸,并释放出游离的FITC分子,在高离子强度环境下(猜想,故而加了氯化镁和钾),与GO之间吸附较小保持相当强的荧光 4. 梭肽酶(carboxypeptidase Y, CPY)、Staurosporine、弗斯可林(Fskorlin)和3-异丁基-1-甲基黄嘿呤(IBMX) 购买于西格玛公司(中国上海)。cAMP-依赖型蛋白激酶(PKA,催化亚基)购买 于Promega公司,酷蛋白激酶II (CKII)购买于New England Biolabs公司(美国)。 突光素标记底物多肽:FITC-RRRADDSDDDDD ( FITC-pep )、 FITC-RRRADDpSDDDDD (FITC-Ppep)和FITC-LRRASLG (FITC-kemptide) ATP、蛋白酶抑制剂和改良型Bradford蛋白总浓度检测试剂盒 牛血清蛋白(BSA)、三轻甲基氨基甲焼(Tris)、甘油、DTT、 EDTASynergy Mx酶标仪(BioTek仪器有限公司)进行突光光谱分析,所有样 品用480 nm作为激发波长,从500 nm到700 nm (25 °C)扫描焚光发射谱图。 5氧化石墨稀与多肽FITC-pep之间的劳光淬灭(氧化石墨烯对荧光强度的影响)FlTC-peptide+ TBS+ Tris-HCl+ MgCb+ KCl+氧化石墨稀 在96孔酶标板中加入60 nL浓度为的多肽(FlTC-peptide)溶液(溶 于TBS,即20 mM Tris-HCl,10 mM MgCb, 50 mM KCl, pH 7.5),每孔中加入
食品中农药残留的检测方法 1 波谱法 该方法是根据有机磷农药中某些官能团或水解、还原产物与特殊的显色剂在特定条件下发生氧化、磺酸化、酯化、络合等化学反应,产生特定波长的颜色反应来进行定性或定量(限量) 测定。 2.色谱法 2.1 薄层色谱法(TLC) 薄层色谱法是一种成熟的、应用也较广的微量快速检测方法。它在农药残留测定技术上有它独特的用处,它既是重要的分离手段,又是定性、定量的分析方法。 检测过程一般先用适宜的溶剂提取有机磷农药,经纯化浓缩后,在薄层硅胶板上分离展开,显色后与标准的有机磷农药比较Rf 值进行定性测定或用仪器进行定量测定。 2.2 气相色谱法(GC) 该方法是利用经提取、纯化、浓缩后的有机磷农药注入气相色谱柱,程序化升温汽化后,不同的有机磷农药在固相中分离,经不同的检测器检测扫描绘出气相色谱图,通过保留时间来定性,通过峰或峰面积与标准曲线对照来定量。一次可同时测定多组份,简便快捷,灵敏度高,准确性也好。而色谱条件的最佳设定是气相色谱技术的关键。 2.3 高效液相色谱法(HPLC) 高效液相色谱法是在液相色谱柱层析的基础上,引入气相色谱理论并加以改进而发展起来的色谱分析方法。高效液相色谱法在农药残留分析的应用越来越广泛,是因为高效液相色谱法能适合分析沸点高而不太容易汽化、热不稳定和强极性农药及其代谢产物;且可以与柱前提取、纯化及柱后荧光衍生化反应和质谱等联用,易实现分析自动化;同时一些新型检测器的问世在一定程度上提高了高效液相色谱法的检测灵敏度。与气相色谱法相比,不仅分离效能好,灵敏度高,检测速度快,而且应用面广。 3 酶抑制法 有机磷农药对哺乳动物中毒作用的基础,通常与它们抑制中枢和周围神经系
有机磷类农药残留量测定法 1 简述 很多有机磷类农药具有毒性,其残留严重危及人体健康。《中国药典》2010年版一部收载了有机磷类农药(对硫磷、甲基对硫磷、乐果、氧化乐果、甲胺磷、久效磷、二嗪农、乙硫磷、马拉硫磷、杀扑磷、敌敌畏、乙酰甲胺磷)的测定方法。 本法通过提取、净化和富集等步骤制备供试品溶液,采用气相色谱法,氮磷检测器测定。 2 仪器与用具 2.1 气相色谱仪,带有氮磷检测器(NPD),载气为高纯氮(纯度>99.9999%)。 2.2 超声仪。 2.3 旋转蒸发仪。 2.4 多功能真空样品处理器(如SUPELCO,isiprep TM DL)。 2.5 活性炭小柱(120~400目,石墨碳填料0.25g,内径0.9cm,3ml)。 2.6 氮吹仪(如Organomation Associates,Inc.,N-EV AP TM 112 nitrogen evaporator)。 2.7 色谱柱:DB-17MS或HP-5弹性石英毛细管柱(30m×0.25mm×0.25μm)或类似极性的毛细管柱。 2.8 具塞锥形瓶、250ml平底烧瓶、棕色量瓶、移液管等。 3 试药与试液 3.1 无水硫酸钠为分析纯。 3.2 乙酸乙酯、正己烷(农残级或分析纯试剂经过全玻璃蒸馏装置重蒸馏,经气相色谱法确认,符合农残检测的要求)。 3.3 农药对照品:对硫磷、甲基对硫磷、乐果、氧化乐果、甲胺磷、久效磷、二嗪农、乙硫磷、马拉硫磷、杀扑磷、敌敌畏、乙酰甲胺磷,由国家标准物质研究中心及农业部环境保护科研检测所提供,其纯度大于99%;也可以使用国际认可的、纯度要求等符合规定的进口标准物质。 4 色谱条件与系统适用性试验 进样口温度:220℃;检测器温度:300℃。不分流进样。程序升温:初始120℃,每fenzh 10℃升至200℃。每分钟5℃升至240℃,保持2min,每分钟20℃升至270℃,保持0.5min。理论板数按敌敌畏峰计算应不低于6000,两个相邻色谱峰的分离度应大于1.5。 5 操作方法 5.1 对照品储备液的制备精密称取对硫磷、甲基对硫磷、乐果、氧化乐果、甲胺磷、久效磷、二嗪农、乙硫磷、马拉硫磷、杀扑磷、敌敌畏、乙酰甲胺磷农药对照品适量,用醋酸乙酯分别制成每1ml约含100μg的溶液,即得。 5.2 混合对照品储备液的制备精密量取上述各对照品储备液1ml,置20ml棕色量瓶中,加乙酸乙酯稀释至刻度,摇匀,即得。 5.3 混合对照品溶液的制备精密量取上述混合对照品储备液,用乙酸乙酯制成每1ml 分别含0.1μg、0.5μg、1μg、2μg、5μg的溶液,即得。 5.4 供试品溶液的制备药材取供试品粉末(过二号筛)约5g,精密称定,加无水硫酸钠5g,加入乙酸乙酯50~100ml,冰浴超声处理3min,放置,取上层液滤过,药渣加乙酸乙酯30~50ml,冰浴超声处理2min,放置,滤过,合并两次滤液,用少量乙酸乙酯洗涤滤纸及残渣,与上述滤液合并。取滤液于40℃下减压浓缩至近干,用乙酸乙酯转移至5ml量瓶中,并稀释至刻度,精密量取1ml,置活性炭小柱[120~400目,0.25g,内径0.9cm(如Supelclean ENVI-Carb SPE Tubes,3ml活性炭小柱),用乙酸乙酯5ml预洗]上,置多功能真空样品处理器上,用正己烷-乙酸乙酯(1:1)的混合溶液5ml洗脱,收集洗脱液,置氮吹仪
分子荧光分析法基本原理 一. 分子荧光的发生过程 (一)分子的激发态——单线激发态和三线激发态 大多数分子含有偶数电子,在基态时,这些电子成对地存在于各个原子或分子轨道中,成对自旋,方向相反,电子净自旋等于零:S=?+(-?)=0,其多重性M=2S+1=1 (M 为磁量子数),因此,分子是抗(反)磁性的,其能级不受外界磁场影响而分裂,称“单线态”; 图1 单线基态(A)、单线激发态(B)和三线激发态(C) 当基态分子的一个成对电子吸收光辐射后,被激发跃迁到能量较高的轨道上,通常它的自旋方向不改变,即 ?S=0,则激发态仍是单线态,即“单线(重)激发态”; 如果电子在跃迁过程中,还伴随着自旋方向的改变,这时便具有两个自旋不配对的电子,电子净自旋不等于零,而等于1: S=1/2+1/2=1 其多重性: M=2S+1=3 即分子在磁场中受到影响而产生能级分裂,这种受激态称为“三线(重)激发态”; “三线激发态” 比“单线激发态” 能量稍低。但由于电子自旋方向的改变在光谱学上一般是禁阻的,即跃迁几率非常小,只相当于单线态→单线态过程的 10-6~10-7。 (二)分子去活化过程及荧光的发生: (一个分子的外层电子能级包括S0(基态)和各激发态S1,S2,…..,T1…..,每个电子能级又包括一系列能量非常接近的振动能级) 处于激发态的分子不稳定,在较短的时间内可通过不同途径释放多余的能量(辐射或非辐射跃迁)回到激态,这个过程称为“去活化过程”,这些途径为: 1. 振动弛豫:在溶液中,处于激发态的溶质分子与溶剂分子间发生碰撞,把一部分能量以热的形式迅速传递给溶剂分子(环境),在10-11~10-13 秒时间回到同一电子激发态的最低振动能级,这一过程称为振动弛豫。
蔬菜中农药残留检测方法研究 【摘要】随着栽培技术的不断进步,农药残留的问题越来越严重,对消费者的身体健康构成了严重威胁。开展蔬菜中农药残留检测方法的研究是控制农药残留保证食品安全的基础,具有重大的意义。本文介绍了蔬菜中农药残留检测的各种方法并对前景进行了展望。 【关键词】蔬菜、农药残留、检测、研究进展 随着栽培技术的不断进步,蔬菜的生长期已越来越短,而随着环境污染的加剧,蔬菜的病虫害也越来越重,绝大部分蔬菜需要连续多次放药后才能成熟上市。农药污染较重的有叶类蔬菜,其中韭菜、油菜受到的污染比例最大。茄果类蔬菜如青椒、番茄等,嫩荚类蔬菜如豆角等,鳞茎类蔬菜如葱、蒜、洋葱等,农药的污染相对较小。农药残留监测体系的建立,对农药残留的监测手段和检测水平提出了更高要求,并促进了农药残留快速检测方法的研究和应用进展,使农药残留检测技术朝着更加快速方便、灵敏可靠的方向发展,逐渐以农药残留专业检测机构的少量检测为中心,向现场检测及实验室的大量检测辐射翻。 1 仪器分析法 由于农药的活性成分大多是小分子有机化合物,故多使用气相色(GC,)~41、高效液相色谱(HPLC,)~、气相色谱一质谱联用(GC-MS)嘲和高效液相色谱一质谱联用(HPLC—Ms)同等技术。其中研究最多的是色质联用技术。因为色质联用特别适合于多种标样残留分析,所以国外把它也划为农药残留快速检测技术之列。大部分农药(如有机氯、有机磷、拟除虫菊酯等)残留可使用GC—MS检测昀,检出限一般为1~10 b~g/kg,但对分子量较大、极性或热不稳定性太强的农药及其化合物,GC-MS不适用,需采用高效液相色谱一质谱联用(HPLC-MS)和其他的方法来检测。 1.1 固相萃取技术 固相萃取法是1种基于液相色谱分离机制的样品制备方法,已广泛应用于农药残留检测工作。它根据液相分离、解析、浓缩等原理,使样品溶液混合物通过柱子后,样品中某一组分保留在柱中,选择合适的溶剂把保留在柱中的组分洗脱下来,从而达到分离、净化的目的。SPE克服了液一液萃取技术及一般柱层析的缺点,具有高效、简便、快速、安全、重复性好、便于前处理自动化等特点。根据柱中填料大体可分为吸附型(如硅胶、大孔吸附树脂等)、分配型(c。,c 、苯基柱等)和离子交换型。1L.R_odriguez等人采用固相萃取法通过改变移动相中缓冲液的浓度、pH值、表面活性剂的浓度和类型对蔬菜中的木精、笨基苯酚、锑比灵和有机磷残留量进行分析,结果表明:pH9.2,缓冲液中含有4mmoUL硼酸和75mmol/L胆酸钠能够得到最好的结果。 1.2 固相微萃取 加拿大Waterloo大学Pawliszyn 1990年首创的一种无需溶剂的萃取技术,它是在固相萃取的基础上发展起来的一种新型的预处理技术。SPME技术由固相萃取技术(SPE)发展而来,对目标化合物有较好的选择性,并且有较高的灵敏度,
阿司匹林片剂的检测 一.阿司匹林体外含量测定 1.两步滴定法测定阿司匹林片的含量: 取阿司匹林片样品10片,精密称定,研磨细,精密称取片粉适量(约相当于阿司匹林0.3g),放在锥形瓶中,加入中性乙醇20mL,振摇,使阿司匹林充分溶解,加 入3滴酚酞指示剂,用氢氧化钠滴定液(0.1mol/L)滴定至溶液呈粉红色。 在上述供试品溶液中,精密加入氢氧化钠滴定液(0.1mol/L)40mL,在水浴上边振摇边加热15min后,迅速放冷至室温,用硫酸迪斯尼工业(0.05mol/L)滴定,并将滴定结果用空白试验进行校正。每1mL的氢氧化钠滴定液(0.1mol/L)相当于18.02mg的C9H8O4(阿司匹林)。 标示量%=(V0-V)*F*T*W平/(W*标示量)*100% V0为空白实验消耗的硫酸滴定液的体积(mL);V为样品测定时消耗的硫酸滴定液的体积(mL);F为硫酸滴定液的浓度校正因子;T为氢氧化钠滴定液的滴定度(mg/mL);W为供试品片粉的取样量(g);W平为供试品的平均片重(g);标示量为片剂的规格(mg/片)。 2.仪器分析法 2.1 Hplc法测定阿司匹林片的含量 色谱条件:ODS Cl8 Hypersil(150 mm*4.6mm,5 um)色谱柱,以水一乙腈一磷酸(76:24:0.5)为流动相;检测波长为276 nm;流速1.3 mL/min;进样量20μL;理论板数不低于10 000。 溶液的制备:供试品制备精密量取样品适量置50 mL容量瓶中,加流动相适量,超声使其溶解,用流动相稀释至刻度(得到200μg/mL的溶液),备用。 对照品的制备:精密称取阿司匹林对照品100mg于50 mL容量瓶中,加流动相适量,超声使其溶解,用流动相稀释至刻度(得2 000μg/mL的溶液)+-,备用。空白溶液制备除阿司匹林外,精取处方量的辅料,配置空白样品溶液。按供试品制备方法制备对照品溶液。通过外标法计算样品浓度。 C=A*Co*D/Ao C为样品浓度(mg/mL);A和Ao分别为样品和对照品溶液的峰面积;D为稀释倍数 (mL)。 2.2 紫外分光光度计法测量阿司匹林片的含量 2.2.1标准溶液系列和样品溶液的配制 分别取0.5g/ml乙酰水杨酸标准溶液0.00mL(空白溶液),0.50mL,1.00mL,1.50mL,2.00mL和阿司匹林样品溶液 1.00mL置于25mL的容量瓶中,分别加入7%
有机磷农药残留的危害及检测方法 姓名:梅增健学号:201008011053 班级:10生工一班 摘要:近年来由于农药的大量使用致使食品的农药残留问题倍受关注,提高农药残留分析检测技术是解决农药残留问题的重要手段。在农药残留检测中样品前处理技术又是检测过程中耗时最长,最容易出现误差的步骤。因而,发展样品前处理技术能提高农药残留检测的效率和准确率。本文综述了近年来食品中农药有机磷残留分析中超临界流体萃取法、固相萃取、固相微萃取和凝胶渗透色谱几种样品前处理技术。 关键词:农药残留;有机磷农药残留;检测;危害 正文: 一、有机磷类农药的危害及特性: 有机磷类农药自问世到现在已有70 年的历史。因为高效、快速、广谱等特点, 有机磷类农药一直在农药中占有很重要的位置, 对世界农业的发展起了很重要的作用。我国已生产和使用的有机磷类农药达数10种之多,其中最常用的有敌百虫、敌敌畏、乐果、马拉硫磷等。但随着这些有机磷类农药的广泛被使用, 暴露出了很多问题,如高残留、毒性强等,尤其在环保意识日益增强的今天,其暴露的问题也引起了人们的高度重视。部分非持久有机磷类农药在某些环境条件下也会有较长的残留期, 并在动物体内产生蓄积。如马拉硫磷是一种高选择性有机磷类农药,在环境中的残留不容忽视,水体中已有检出。马拉硫磷对水生生物属高毒农药, 对人免疫功能也具有一定的毒性作用, 已成为水环境中重要的监测项目。大多数有机磷类农药都属于磷酸酯类或硫代磷酸酯类化合物,其中有机磷酸脂类化合物纯品多为油状,少数为结晶固体。常用剂型有乳剂、油剂、粉剂及颗粒剂等。有机磷类农药的中毒特征是血液中胆碱酯酶活性下降, 胆碱酯酶的活性受到抑制,导致神经系统机能失调,从而使一些受神经系统支配的脏器,如心脏、支气管、肠、胃等发生功能异常。 二、有机磷农药的检测前处理 1、超临界萃取技术 所谓超临界是物质的一种特殊流体状态:当气液平衡的物质升温升压时,热膨胀会引起液体密度减少,压力升高又会使气相密度变大,当温度和压力达到某一点时,气液两相界面消失成一均相体系,此点即为临界点;当物质的温度和压力高于临界点时,就处于超临界状态,此时该物质为超临界流体。超临界萃取技术(supercritical fluid extraction,SFE)是用超临界液体作为萃取剂,根据样品中组分在不同压力和温度条件下溶解能力变化的性质,通过改变萃取剂流体的压力,从而将不同组分萃取出来。 等流体代替有机溶剂,对人体无害、绿色环保,且SFE最大优点是使用CO 2 有效地防止了热敏性物质的氧化和逸散,提高了样品回收率,因而较适合于萃取热不稳定物质和有毒化合物[1]。D.H.Kim等[2]已将SFE用于萃取面粉里的有机磷残留,避免了样品浓缩和分析过程的干扰,提高了萃取速度,60min有7g的样品萃取量,且联用GC-NPD分析后,检测限低至10ng/s。
分子荧光光谱法实验报告范文 一、实验目的 1.掌握荧光光度计的基本原理及使用。 2.了解荧光分光光度计的构造和各组成部分的作用。 3.掌握分子荧光光度计分析物质的特征荧光光谱:激发光谱、发射光谱的测定方法。 4.了解影响荧光产生的几个主要因素。 5.学会运用分子荧光光谱法对物质进行定性和定量分析。 二、实验原理 原子外层电子吸收光子后,由基态跃迁到激发态,再回到较低能级或者基态时,发射出一定波长的辐射,称为原子荧光。对于分子的能级激发态称为分子荧光,平时所说的荧光指分子荧光。 具有不饱和基团的基态分子经光照射后,价电子跃迁产生荧光,是当电子从第一激发单重态S1的最低振动能级回到基态S0各振动能级所产生的光辐射。 (1)激发光谱 是指发光的某一谱线或谱带的强度随激发光波长(或频率)变化的曲线。横坐标为激发光波长,纵坐标为发光相对强度。 激发光谱反映不同波长的光激发材料产生发光的效果。即表示发光的某一谱线或谱带可以被什么波长的光激发、激发的本领是高还是低;也表示用不同波长的光激发材料时,
使材料发出某一波长光的效率。荧光为光致发光,合适的激发光波长需根据激发光谱确定——激发光谱是在固定荧光波长下,测量荧光体的荧光强度随激发波长变化的光谱。获得方法:先把第二单色器的波长固定,使测定的λem不变,改变第一单色器波长,让不同波长的光照在荧光物质上,测定它的荧光强度,以I为纵坐标,λex为横坐标所得图谱即荧光物质的激发光谱,从曲线上找出λex,,实际上选波长较长的高波长峰。 (2)发射光谱 是指发光的能量按波长或频率的分布。通常实验测量的是发光的相对能量。发射光谱中,横坐标为波长(或频率),纵坐标为发光相对强度。 发射光谱常分为带谱和线谱,有时也会出现既有带谱、又有线谱的情况。发射光谱的获得方法:先把第一单色器的波长固定,使激发的λex不变,改变第二单色器波长,让不同波长的光扫描,测定它的发光强度,以I为纵坐标,λem为横坐标得图谱即荧光物质的发射光谱;从曲线上找出最大的λem。 (3)荧光强度与荧光物质浓度的关系 用强度为I0的入射光,照射到液池内的荧光物质时,产生荧光,荧光强度If用仪器测得,在荧光浓度很稀(A0.05)时,荧光物质发射的荧光强度If与浓度有下面的关系:If=KC。 三、实验试剂和仪器
附录ⅨQ.农药残留量测定法 本法系用气相色谱法(附录ⅥE)测定药材和饮片及制剂中部分有机氯、有机磷和拟除虫菊酯类农药,除另有规定外,按下列方法测定。 一、有机氯类农药残留量测定 色谱条件与系统适用性试验弹性石英毛细管柱(30m×0.32mm×0.25μm) SE-54,63Ni-ECD电子捕获检测器。进样口温度230℃;检测器温度300℃。不分流进样。程序升温:初始100℃,每分钟10℃升至220℃,每分钟8℃升至250℃,保持10分钟。理论板数按α-BHC峰计算应不低于1×106,两个相邻色谱峰的分离度应大于1.5。 对照品储备液制备精密称取六六六(BHC)[α-BHC,β-BHC,γ-BHC,δ-BHC),滴滴涕(DDT)[ PP’-DDE,PP’-DDD,OP’-DDT,PP’-DDT]及五氯硝基苯(PCNB)农药对照品适量,用石油醚(60~90℃)分别制成每1ml约含4~5μg的溶液,即得。 混合对照品储备液的制备精密量取上述各对照品储备液0.5ml置10ml量瓶中,用石油醚(60~90℃)稀释至刻度,即得。 混合对照品溶液的制备精密量取上述混合对照品储备液,用石油醚(60~90℃)制成每1L含0μg、1μg、5μg、10μg、50μg、100μg、250μg的溶液,即得。 供试品溶液制备药材和饮片取供试品于60℃干燥4小时,粉碎成细粉,取约2g,精密称定,置100ml具塞锥形瓶中,加水20ml浸泡过夜,精密加丙酮40ml,称定重量,超声处理30分钟,放冷,再称定重量,用丙酮补足减失的重量,再加氯化钠约6g及二氯甲烷30ml,称定重量,超声处理15分钟,再称定重量,用二氯甲烷补足减失的重量,静置(使分层),将有机相迅速移入装有适量无水硫酸钠的100ml具塞锥形瓶中,放置4小时。精密量取35ml,于40℃水浴上减压浓缩至近干,加少量石油醚(60~90℃)如前反复操作至二氯甲烷及丙酮除净,用石油醚(60~90℃)溶解并转移至10ml具塞刻度离心管中,加石油醚(60~90℃)至5ml。小心加入硫酸1ml,振摇1分钟,离心(3000转/分)10分钟。精密量取上清液2ml置具刻度的浓缩瓶中,连接旋转蒸发器,40℃下(或用氮气)将溶液浓缩至适量、精密稀释至1ml,即得。 制剂取供试品,研成细粉(蜜丸切碎,液体制剂直接量取),精密称取适量(相当于药材和饮片2g),以下按上述供试品溶液制备,即得供试品溶液。 测定法分别精密吸取供试品溶液和与之相对应浓度的混合对照品溶液各1μl,分别连续进样3次,取3次平均值,按外标法计算供试品中9种农药残留量。 二、有机磷类农药残留量测定
1.主题内容:建立有农药残留量测定法操作方法。 2.适用范围:本规程适用于检查药物在生产过程中的农药残留量测定法的操作。 3.引用标准:《中国药典2010版一部》 4.责任:化验员、QC主管。 5. 用途:化验室 6.检查内容及方法 本法系用气相色谱法(附录ⅥE)测定药材、饮片及制剂中部分有机氯、有机磷和拟除虫菊酯类农药,除另有规定外,按下列方法测定。 6.1有机氯类农药残留量测定 6.1.1色谱条件与系统适用性试验 弹性石英毛细管柱(30m×0.32mm×0.25μm)SE-54(或DB-1701),63Ni-ECD电子捕获检测器。进样口温度230℃,检测器温度300℃,不分流进样。程序升温:初始100℃,每分钟10℃升至220℃,每分钟8℃升至250℃,保持10分钟,理论板数按α-BHC峰计算应不低于1×106,两个相邻色谱峰的分离度应大于1.5. 6.1.2对照品储备液的制备 精密称取六六六(BHC)(α-BHC,β-BHC,γ-BHC,δ-BHC)、滴滴涕(DDT)(PP′-DDE,PP′-DDD,OP′-DDT,PP′-DDT)及五氯硝基苯(PCNB)农药对照品适量,用石油醚(60~90℃)分别制成每1ml约含4~5μg的溶液,即得。 6.1.3混合对照品储备液的制备 精密量取上述各对照品储备液0.5ml,置10ml量瓶中,用石油醚(60~90℃)稀释至刻度,摇匀,即得。 6.1.4混合对照品溶液的制备 精密量取上述各对照品储备液,用石油醚(60~90℃)制成每1L分别含0μg、1μg、5μ
g、10μg、50μg、100μg、250μg的溶液,即得。 6.1.5供试品溶液的制备 药材或饮片:取供试品于60℃干燥4小时,粉碎成细粉,取约2g,精密称定,置100ml 具塞锥形瓶中,加水20ml浸泡过夜,精密加丙酮40ml,称定重量,超声处理30分钟,放冷,再称定重量,用丙酮不足减失的重量,再加氯化钠约6g,精密加二氯甲烷30ml,称定重量,超声处理15分钟,再称定重量,用二氯甲烷补足减失的重量,静置(使分层),将有机相迅速移入装有适量无水硫酸钠的100ml具塞锥形瓶中,放置4小时。精密量取35ml,于40℃水浴上减压浓缩至近干,加少量石油醚(69~90℃)如前反复操作至二氯甲烷及丙酮除净,再用石油醚(69~90℃)溶解并转移至10ml具塞刻度离心管中,加石油醚(69~90℃)精密稀释至5ml,小心加入硫酸1ml,振摇1分钟,离心(3000转/分)10分钟,精密量取上清液2ml,至具刻度的浓缩瓶(见图)中,连接旋转蒸发器,40℃下(或用氮气)将溶液浓缩至适量,精密稀释至1ml,即得。 6.1.6制剂 取供试品,研成细粉(蜜丸切碎,液体直接量取),精密称取适量(相当于药材2g),以下按上述供试品溶液制备法制备,即得供试品溶液。 6.1.7测定法 分别精密吸取供试品溶液和与之相对应浓度的混合对照品溶液各1μl,分别连续进样3次,取3次平均值,按外标法计算公式品中9中有机氯农药残留量。 6.2有机磷类农药残留量测定 6.2.1色谱条件与系统适用性试验 弹性石英毛细管柱(30m×0.25mm×0.25μm)DB-17MS(或HP-5),磷酸检测器(NPD)。进样口温度220℃,检测器温度300℃,不分流进样。程序升温:初始120℃,每分钟10℃升
荧光光谱法在生物分析中的应用 ---亚甲基蓝与蛋白质的结合反应 刘超-生物化工-2009010236 荧光光谱法是研究生物大分子与小分子、离子相互作用的重要手段。荧光测试中的发射峰特征、荧光偏振、能量转移及荧光寿命等指标可以对蛋白质分子中荧光生色基团的结构及其所处的微环境提供有用的信息。在生物体中,蛋白质是必不可少的生命物质,有关蛋白质的各类研究,是目前生命科学、化学和临床医学中共同关注和感兴趣的课题。亚甲基蓝是一种具有平面结构(结构式见图1)的碱性生物染色剂,在医学临床诊断及化学分析中已有较长的应用历史,且可用于亚硝酸盐、磺氨类、氰化物及一氧化碳等中毒的解毒药。近年来又发现MB对DNA具有插入作用,MB可被用于抗癌药物的体外筛选;此外MB的光化学性质及作用机理与血卟啉很相似,被认为可能成为一种新型光敏剂而用于癌症的光化学治疗。新近的研究还表明MB在溶液中分子的聚集状态与溶剂及介质有关。本文应用荧光光谱法研究了人体生理pH值条件下,亚甲基蓝与牛血清白蛋白间的相互作用,测定了结合常数、结合位点数及结合距离,从而由分子水平的角度认识蛋白质分子与小分子之间相互作用的机理,为生命科学研究提供有用的信息和数据。 实验方法:配制0.05mol/L的Tris-HCl并含0.10mol/LNaCl的缓冲溶液 (pH=7.0),恒定体系pH值和离子强度,以此缓冲溶液分别配制MB溶液和BSA贮备液。测定方法:在10ml的容量瓶中,依此加入一定量的BSA,MB溶液,以缓冲溶液稀释至刻度,混匀后室温下测定荧光光谱、同步荧光光谱及与蛋白质分子比为1:1的MB吸收光谱,测定中固定BSA的浓度而改变MB浓度。 结果与讨论:MB对BSA荧光猝灭的机理外加分子与荧光体分子相互作用引起荧光强度降低的现象称为荧光猝灭,并有动态和静态猝灭之分。通过观察MB对BSA 猝灭曲线(图2)可知,对于MB 从低到高整个浓度范围内曲线都呈现出好的线性,表明室温下MB 对BSA 的荧光猝灭为静态过程。为进一步证实此猝灭特征,把此过程按动态猝灭处理,即式(1) 为双分子猝灭过程速率常数,为动态猝灭常称为Stern-Volmer猝灭方程,K Q 数,为猝灭剂不存在时荧光体分子平均寿命,猝灭剂的浓度。而 ,由于生物大分子荧光平均寿命约为,故可由猝灭曲线的斜率求得猝灭常数。由图2实验数据,按式(1)可以求得MB对BSA 猝灭的,而各类猝灭剂对生物大分子的最大扩散碰撞猝灭常数为,显然MB对BSA荧光的猝灭过程速率常数远大于扩散控制的,可见其猝灭过程确
荧光光度法测定阿司匹林中乙酰水杨酸的含量 一、实验目的 1.掌握用荧光法测定药物中的乙酰水杨酸含量的方法。 2.掌握970CRT 型荧光分光光度计的操作方法。 3.加深对荧光光度法原理的理解。 二、实验原理 1.荧光光度法原理 (1)常温下,处于基态的分子吸收一定的紫外可见光的辐射能成为激发态分子,激发态分子通过无辐射跃迁至第一激发态的最低振动能级,再以辐射跃迁的形式回到基态,发出比吸收光波长长的光而产生荧光。在稀溶液中,当实验条件一定时,荧光强度I F 与物质的浓度c 成线性关系: 即Kc I F (这是荧光光谱法定量分析的理论依据)。 (2)荧光光谱 激发光谱:固定测量波长(选最大发射波长),化合物发射的荧光强度与照射光波长的 关系曲线。激发光谱曲线的最高处,处于激发态的分子最多,荧光强度最大。 发射光谱:固定激发光波长(选最大激发波长), 化合物发射的荧光强度与发射光波长关系曲线。 固定发射光波长进行激发光波长扫描,找出最大激发光波长,然后固定激发光波长进 荧 光强度 波长
行荧光发射波长扫描,找出最大荧光发射波长。激发光波长和发射荧光波长的选择是本实验的关键。 2. 荧光光度法测定阿司匹林中乙酰水杨酸的含量 通常称为ASA 的乙酰水杨酸(阿司匹林)水解即生成水杨酸(SA )(如下式)。而在阿司匹林中或多或少存在一些水杨酸,用醋酸—氯仿作为溶剂,然后用荧光法可以分别测定其含量,少许醋酸还可以增加二者的荧光强度(本次实验只测定阿司匹林中乙酰水杨酸的含量)。在1%的乙酸—氯仿中乙酰水杨酸的激发光谱和荧光光谱如图所示:(为了消除药片之间的差异,可以取几片一起研磨,然后取部分由代表性的样品进行分析) 三、仪器与试剂: 仪器:970CRT 型荧光分光光度计及附件;容量瓶:1000mL 2只,100 mL 2只,50mL 8只;l0mL 吸管2支;铁架台;研钵;称量瓶;玻璃棒;烧杯;定量滤纸;电子天平。 试剂:冰醋酸;氯仿;乙酰水杨酸;阿司匹林;丙酮。 四、实验步骤: 1. 接通电源,打开氙灯,再打开主机,然后打开计算机启动工作站并初始化仪器,预热30min 左右。 2. 仪器初始化完毕后,在工作界面上选择测量项目,设置适当的仪器参数。 3.配置溶液: (1)配置1%的醋酸—氯仿溶剂:在1000ml 容量瓶中,将冰醋酸与氯仿以1:99的比例配制。 (2)配置储备液:准确称取0.4000g 乙酰水杨酸溶于1%的醋酸—氯仿溶剂中制成溶液,在1000ml 的容量瓶中定容备用。(400ug|ml ) (3)配置4.00 ug|ml 的ASA 溶液:在100 ml 容量瓶中将(2)中的标准液稀释100倍.(扫光谱) (CH 3CO)2O H 2O (ASA) (SA)