
达比加群酯合成路线综述
一、 合成路线
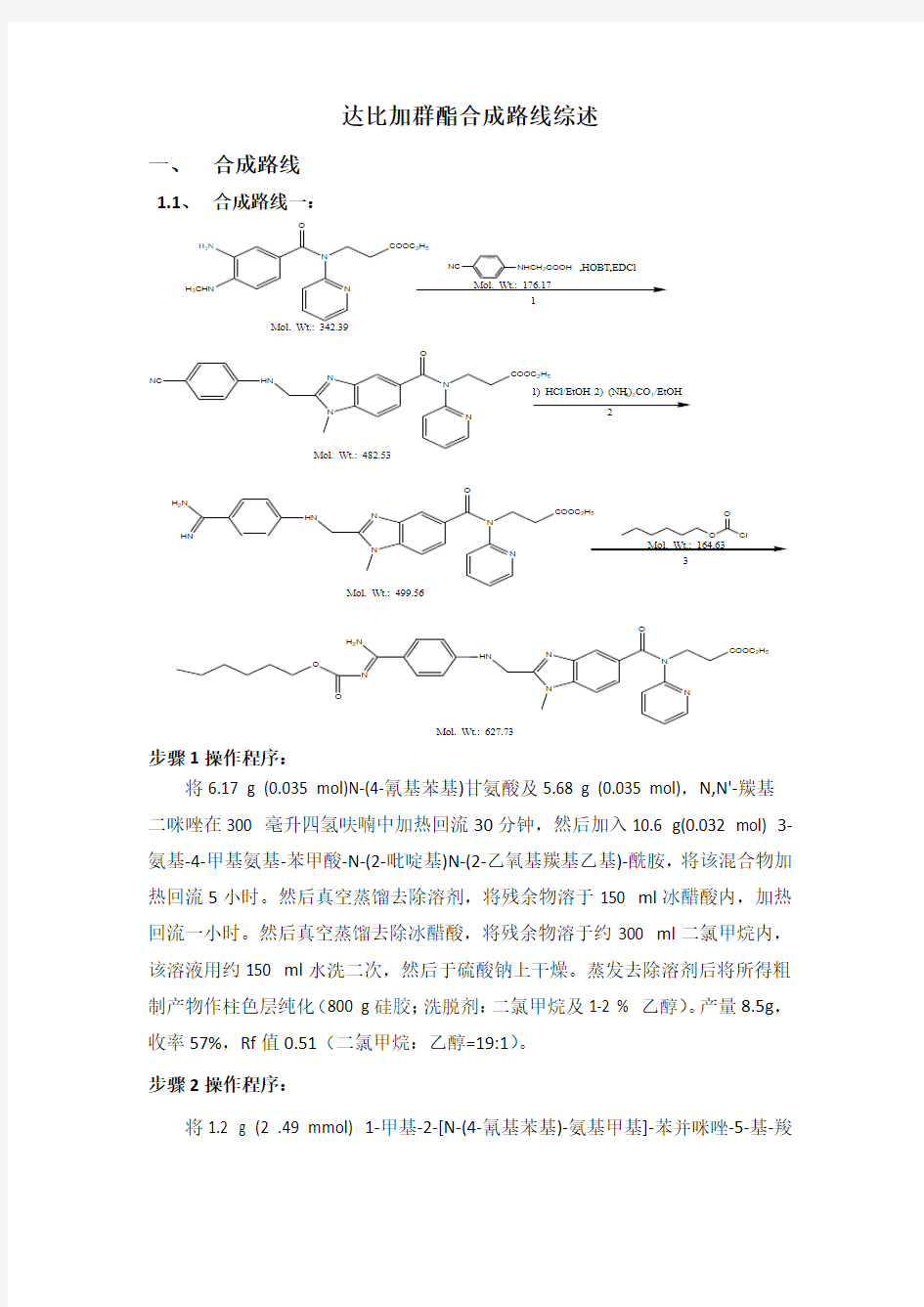
1.1、 合成路线一:
N
COOC 2H 5
H 3CHN
H 2N
N
O
NC
NHCH 2COOH ,HOBT,EDCl
N
COOC 2H 5
N
O
N
N HN
NC N
COOC 2H 5
N
O
N
N
HN
HN
H 2N
O
O 1
3
N
COOC 2H 5
N
O
N
N
HN
N
H 2N
O
O
Mol. Wt.: 627.73
Mol. Wt.: 164.63
Mol. Wt.: 499.56
Mol. Wt.: 482.53
Mol. Wt.: 176.17
Mol. Wt.: 342.39
1) HCl/EtOH 2) (NH 4)2CO 3/EtOH
步骤1操作程序:
将6.17 g (0.035 mol)N-(4-氰基苯基)甘氨酸及5.68 g (0.035 mol),N,N'-羰基 二咪唑在300 毫升四氢呋喃中加热回流30分钟,然后加入10.6 g(0.032 mol) 3-氨基-4-甲基氨基-苯甲酸-N-(2-吡啶基)N-(2-乙氧基羰基乙基)-酰胺,将该混合物加热回流5小时。然后真空蒸馏去除溶剂,将残余物溶于150 ml 冰醋酸内,加热回流一小时。然后真空蒸馏去除冰醋酸,将残余物溶于约300 ml 二氯甲烷内,该溶液用约150 ml 水洗二次,然后于硫酸钠上干燥。蒸发去除溶剂后将所得粗制产物作柱色层纯化(800 g 硅胶;洗脱剂:二氯甲烷及1-2 % 乙醇)。产量8.5g ,收率57%,Rf 值0.51(二氯甲烷:乙醇=19:1)。 步骤2操作程序:
将1.2 g (2 .49 mmol) 1-甲基-2-[N-(4-氰基苯基)-氨基甲基]-苯并咪唑-5-基-羧
酸-N-(2-吡啶基)-N-(2- 乙氧基羟基乙基)-酰胺在100 毫升饱和盐酸的乙醇溶液中在室温搅拌6 小时.将该混合物在真空蒸发至干,残余物溶在100 ml乙醇中与2.5 g(26 mmol-)碳酸铵混合,在室温下搅拌过夜.经蒸馏去除溶剂后,将所得粗制产物进行柱色层纯化(100 g硅胶;洗脱剂;二氯甲烷/乙醇=4:1)。将洗脱液浓缩后得所需化合物,为白色固体。产量1. 10 g,收率83%,Rf值0.18(二氯甲烷/乙醇=4:1)。
步骤3操作程序:
将l.l g(2.06mmol) 1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2- 乙氧基羟基乙基)-酰胺盐酸盐溶于由40 ml四氢呋喃和10 ml 水构成的混合物内,然后加入570 mg(4.12 mmol)碳酸钾和362 mg(2.2 mmol)氯甲酸正己酯,于室温搅拌二小时,然后浓缩蒸去溶剂,残余物与约50 毫升饱和盐水溶液混合,所得溶液用每次用20ml二氯甲烷萃取三次.将萃取液在硫酸钠上干燥,蒸馏得粗产物进行柱色层纯化(1 00克硅胶;二氯甲烷+ 5 %乙醇)。产量0.66 g,收率51%,Rf值0.53(二氯甲烷/甲醇=9:1)。
1.2、 合成路线二:
OH
O
N
N
H N
NC
222
THF
N
HN
O
O
N
2H 5
N
O
N
N HN
NC 1) HCl/EtOH 2) (NH 4)2CO 3/EtOH
N
COOC 2H 5
N
O
N
N
HN
HN
H 2N
O
O 3
N
COOC 2H 5
N
O
N
N
HN
N
H 2N
O
O
Mol. Wt.: 627.73
Mol. Wt.: 164.63
Mol. Wt.: 499.56
Mol. Wt.: 482.53
1
Mol. Wt.: 306.32
Mol. Wt.: 194.23
步骤1操作程序:
将2.0 g(6.5 mmol)3-甲基-2-[2-(4-氰基苯基)乙基]-咪唑并[4,5-b]吡啶-6-羧酸在100 ml 二氯甲烷中的溶液和20 ml 氯化亚砜混合,回流2 小时.待蒸馏掉液体成分后,将粗制产物溶于二氯甲烷中二次,每次都蒸馏掉溶剂。将这样制得酰基氯(2 g)悬浮于l00 ml 四氢呋喃中,与1.2 g(6.5 mmol) N-2吡啶-B-丙氨酸乙酯混合.然后用5分钟滴加0.73 g(7.2 mmol)三乙胺,揽拌半小时后,真空蒸馏掉溶剂,将残余物溶于乙酸乙醋中,有机相用水洗涤,用硫酸钠干燥.浓缩掉溶剂后过柱(硅胶;二氯甲烷至二氯甲烷/乙醇= 49:1)后,分离出所需产物,为棕色油体。1.9 g,收率65%,Rf 值:0.44(乙酸乙酯/乙醇/氨水=90:10:1)。 步骤2操作程序:
将1. 8 g(3.7 mmol) 1-甲基-2-[N-(4-氰基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2- 乙氧基羟基乙基)-酰胺加入到100 ml 氯化氢饱和乙醇溶液中搅拌16 小时,先于0℃它搅拌,再于室温搅拌直至以TLC 点板测不出有起始物
料。蒸馏掉溶剂,将油状产物溶于50 ml无水乙醇中,加入3.6 g(37 mmol)碳酸铵,经4小时后,真空蒸馏掉溶剂,所得的粗品过柱(硅胶;梯度;二氯甲烷/乙醇19:1至4:1),产量1.6 g,收率80%,Rf值:0.3(乙酸乙酯/乙醇/氨水=90:5:5 步骤3操作程序:
将l.l g(2.06mmol) 1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2- 乙氧基羟基乙基)-酰胺盐酸盐溶于由40 ml四氢呋喃和10 ml 水构成的混合物内,然后加入570 mg(4.12 mmol)碳酸钾和362 mg(2.2 mmol)氯甲酸正己酯,于室温搅拌二小时,然后浓缩蒸去溶剂,残余物与约50 毫升饱和盐水溶液混合,所得溶液用每次用20ml二氯甲烷萃取三次.将萃取液在硫酸钠上干燥,蒸馏得粗产物进行柱色层纯化(1 00克硅胶;二氯甲烷+ 5 %乙醇)。产量0.66 g,收率51%,Rf值0.53(二氯甲烷/甲醇=9:1)。
1.3、 合成路线三:
H N
HO
O
N HN
O O
N
2H 5
H 3CHN H 2N
N
O
N
COOC 2H 5
N
O
N
N
HN
N
O
N H
O
Mol. Wt.: 342.39
Mol. Wt.: 235.2
Mol. Wt.: 541.56
1.PPA/THF 或CDI/THF
2.AcOH/EtOH 或AcOH/EE
H Pd/C
THF, H 2O,ACOH
或PTSA
N
COOC 2H 5
N
O
N
N
HN
HN
H 2N
O
O 3
N
COOC 2H 5
N
O
N
N
HN
N
H 2N
O
O
Mol. Wt.: 627.73
Mol. Wt.: 164.63
Mol. Wt.: 499.56
1
2
N
COOC 2H 5
N
O
N
N
HN
N H 2N
O
O
.CH 3SO 3H
C 35H 45N 7O 8S Mol. Wt.: 723.84
S OH
O
O
H 3C
4
步骤1操作程序1:
将11.35 g(70mmol)1, 1'-羰基二咪唑悬浮于100 mlTHF 中且加热至50℃。分批添加14.23g( 60.5mmol) 2-[4-(1,2,4-噁二唑-5-酮-3-基)-苯氨基]-乙酸。将17.1g(50mmol) 3-氨基-4-甲基氨基-苯甲酸-N-(2-吡啶基)N-(2-乙氧基羰基乙基)-酰胺加入到37ml 四氢呋喃中,并在50℃加热下溶解。约90min 之后,将2-[4-(1,2,4-噁二唑-5-酮-3-基)-苯氨基]-乙酸悬浮液计量添加至3-氨基-4-甲
基氨基-苯甲酸-N-(2-吡啶基)N-(2-乙氧基羰基乙基)-酰胺溶液中,且以20ml四氢呋喃冲洗。将该反应混合物搅拌约18h ,且接着在添加100ml乙酸后加热回流,以使四氢呋喃蒸馏掉。约lh 后,添加400ml水且搅拌该混合物。将该溶液冷却,将所沉淀的粉红色固体物质滤出且以20ml 水分2 次洗涤并于真空下在最大50℃下干燥。经分离的物质为(3) 的二乙酸盐。产量24.8g (收率75%); 熔点:167℃,纯度>95%HPLC 峰面积。
步骤1操作程序2:
将34.2g(O.lmol)3-氨基-4-甲基氨基-苯甲酸-N-(2-吡啶基)N-(2-乙氧基羰基乙基)-酰胺、27.5g(O.12mol) 2-[4-(1,2,4-噁二唑-5-酮-3-基)-苯氨基]-乙酸及30 .3 g(O.23mol)二异丙基乙胺置于170ml四氢呋喃中且冷却至稍低于周围温度。接着计量添加85g(0.13mol)丙烷磷酸酐(乙酸乙酯中约50%的溶液)。将该混合物再搅拌90分钟且接着将溶剂蒸馏掉。接近终点时添加73.5g 乙酸且将该混合物加热至90℃的内部温度。接着添加400ml乙醇或优选400ml乙醇/水(约85: 15)且将该混合物热过滤。将该溶液冷却,将沉淀的固体物质滤出且以50ml乙醇分2 次洗涤及于真空下在最大50℃下干燥。经分离的物质为(3) 的二乙酸盐。产量56g (收率75%); 熔点:167℃,纯度>95%HPLC 峰面积。
步骤1操作程序3:
于0℃下,将96g(0 .41mol) 2-[4-(1,2,4-噁二唑-5-酮-3-基)-苯氨基]-乙酸悬浮于250mlN-甲基吡咯烷酮及550ml四氢呋喃中。继而将该稀的悬浮液与48g(0 .4mol)三甲基乙酰基氯及52g(0 .4mol)二异丙基乙胺混合且搅拌30 分钟。接着添加溶解于800ml乙酸中的125g(0.36mol) 3-氨基-4-甲基氨基-苯甲酸-N-(2-
吡啶基)N-(2-乙氧基羰基乙基)-酰胺,且将该反应混合物加热回流3h。在轻微真空下将四氢呋喃蒸馏掉且于温热时计量添加1600ml水。将该固体于5℃下分离,以550ml水洗涤并于循环空气干燥器中在最大50℃下干燥过夜。
步骤2操作程序1:
将37.3g(56.4mmol)1-甲基-2-[N-[4-(1,2,4-噁二唑-5-酮-3-基)-苯基]-氨基-甲基]-苯并咪唑-5-基羧酸-N-(2-吡啶基)-N-(2-乙氧基羰基乙基)-酰胺溶解于900ml乙醇中,且在添加10ml乙酸后,于室温下及2巴氢气下以4g 经水
潮湿的10%Pd/C 氢化。将催化剂滤出且将溶解于180ml乙醇中的17g(89 .4mmol)对甲苯磺酸添加至该滤液。将的1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2- 乙氧基羟基乙基)-酰胺的甲苯磺酸盐沉淀出,滤出且再次以150ml 乙醇分多次洗涤。获得潮湿物质,将其于35℃真空下干燥。产量34.5g ,浅米色物质(收率91.3%),熔点187℃,纯度:>98%HPLC峰面积。
步骤2操作程序2:
将37.3g(56 .4mmol)溶解于1-甲基-2-[N-[4-(1,2,4-噁二唑-5-酮-3-基)-苯基]-氨基-甲基]-苯并咪唑-5-基羧酸-N-(2-吡啶基)-N-(2-乙氧基羰基乙基)-酰胺400ml 乙醇/水(90:10) 中,且室温于及2巴氢气下,以4g 经水潮湿的10%Pd/C 氢化。将催化剂过滤且将11.5g(60.6mmol)对甲苯磺酸添加至该滤液。经蒸发使(4)的甲苯磺酸盐沉淀出。将该悬浮液冷却,将该物质滤出且以150ml 乙醇/水分多次洗涤。获得潮湿物质,将其于35℃真空下干燥。产量33.7g ,浅米色物质(收率89%),熔点187℃,纯度:>98%HPLC峰面积。
步骤2操作程序3:
室温下,将30.0g(45 .3 mmol)1-甲基-2-[N-[4-(1,2,4-噁二唑-5-酮-3-基)-苯基]-氨基-甲基]-苯并咪唑-5-基羧酸-N-(2-吡啶基)-N-(2-乙氧基羰基乙基)-酰胺溶解于90ml THF/水(1: 1) 中,与4g 经水潮湿的10%Pd/C 混合且于4巴氢气下,60℃下氢化。将催化剂滤出,再以大约40ml 的THF/水(l: 1)洗涤且将滤液无需处理用于下一步骤,或如上文所述通过添加溶解于100ml 水中的13.6g(72mmol)对甲苯磺酸进行分离并冷却。
步骤3操作程序1:
在有34g(246mmol)碳酸钾存在下,在约15℃的温度下,将溶解于437ml丙酮及273ml 水中的55g(81.9mmol) 1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2- 乙氧基羟基乙基)-酰胺与16.4g(99.6mmol)氯甲酸己酯混合。反应结束后,将析出的产物滤出且以丙酮/水洗涤。必要时,可将其在加热下再次溶解在约270ml 丙酮中,且接着过滤。过滤后,通过添加220ml 水使该物质再次结晶。将分离的物质于45℃真空下干燥。产量42-48g,收率82-94%。步骤3操作程序2:
在有34g(246mmol)碳酸钾存在下,在约15℃的温度下,将溶解于437ml丙酮及273ml 水中的55g(81.9mmol) 1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2- 乙氧基羟基乙基)-酰胺与16.4g(99.6mmol)氯甲酸己酯混合。反应结束后,将该悬浮液加热至约50℃。分离有机相且由440ml乙酸乙酯代替丙酮。将接着分离的水相丢弃且将有机相以稀释的碳酸钾溶液多次洗涤并最后以水洗涤。将产物冷却析晶,分离及以乙酸乙酯洗涤。45℃真空下干燥。产量42-48g,收率82-94%。
步骤4操作程序:
将l00g(O.16mol) 1-甲基-2-[N-[4-(N-正己氧基羰基甲脒基)苯基]-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2- 乙氧基羟基乙基)-酰胺在加热下溶解于890ml 丙酮中且与15g(O.16mol) 甲磺酸于200ml 丙酮中的溶液混合。将该溶液过滤,加77ml丙酮后冷却至约20℃。将析出的产物分离且以丙酮同再次洗涤。50℃以下真空下干燥。产量103-113,收率90-98%。
1.4、 合成路线四:
N
C 2H 5OOC
N
N
H N
CN
EtOH/HCl (NH 4)2CO 3,CaCl 2
N
C 2H 5OOC
N
N
N
H NH
NH 2
Mol. Wt.: 482.53
N
N
O
N
N
N
N
O
OC 5H 13
N
C 2H 5OOC
N
O
N
H N
NH 2
O
O
Mol. Wt.: 627.73
2
3
N
COOC 2H 5
N
O
N
N
HN
N H 2N
O
O
CH 3
SO 3
H
C 35H 45N 7O 8S Mol. Wt.: 723.84
S OH
O
O
H 3C
4
2. HOOCCOOH
1.
COOH COOH
.
.Mol. Wt.: 589.6
步骤1操作程序:
Calcium chloride dihydrate (12.5g) was added to a mixture of l-methyl-2-[N-(4-cyanophenyl)aminomethyl]benzimidazol-5-ylcarboxylicacid-N-(2-p yridyl)-N-(2-ethoxy carbonyl ethyl)amide compound of formula-14 (50g) and ethanol (750 ml) and stirred for 20 minutes. The reaction mixture was cooled to 0-5℃. and HCl gas was passed into the reaction mixture over a period of 5 hours at a temperature below 10℃. The temperature of the reaction mixture was raised to 25-30. and stirred for 8 hours at the same temperature. After completion of the reaction, the solvent was expelled out under N 2 pressure. The reaction mixture was cooled to 0-5℃. and slowly added ammonium formate (150g). The reaction mixture was stirred for 30 minutes and ammonium carbonate (300 g) was added. The temperature of the reaction mixture
was raised to 25-35℃. and stirred for 10 hours. After completion of the reaction, the reaction mixture was filtered and the filtrate was distilled under reduced pressure. A solution of 10% ethanol in ethyl acetate was added to the reaction mixture and stirred for 3 hours to obtain a solid. Filtered the obtained solid, washed with ethyl acetate and then dried to get the title compound. Yield: 45 g.
A mixture of 1-methyl-2-[N-[4-amidinophenyl]aminomethyl]benzimidazol-5-yl -carboxylicacid-N-(2-pyridyl)-N-(2-ethoxycarbonylethyl) amide compound of formula-5 (100 g) and ethanol (1200 ml) was heated to 50-60℃.A solution of oxalic acid (25.25 g) in ethanol (1500 ml) was added to the above reaction mixture at 50-60℃. and stirred for 45 minutes. The reaction mixture was cooled to 25-35℃. and stirred for 6 hours at 25-35℃. Filtered the solid, washed with ethanol and then dried to get the title compound.
室温下,在50 g 1-甲基-2-[N-(4-氰基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2- 乙氧基羟基乙基)-酰胺中加入乙醇750ml,加入CaCl2 12.5 g,搅拌20min后降温至0-5℃。控温10℃以下,通入HCl气体搅拌反应5h。升温
吹干乙醇。降温至0-5℃,缓慢至25-30℃,搅拌反应8h。反应完成后,通入N
2
加入甲酸铵150g。在30min内加入碳酸铵300g。升温至25-35℃,搅拌反应10h。反应完成后,将反应混合物过滤,将滤液在减压下蒸馏。10%乙醇的乙酸乙酯溶液加入到反应混合物中,并搅拌3小时,得到固体。过滤得到的固体,用乙酸乙酯洗涤,然后干燥,得到目标化合物。产量:45g。
在50g 1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2- 乙氧基羟基乙基)-酰胺中加入1200乙醇,升温至50-60℃,滴加25.25g 草酸加1500乙醇溶液,滴加完毕后搅拌45min。降温至25-35℃搅拌6h。过滤,滤饼用乙醇洗涤,真空干燥的产品。
步骤2操作程序:
A solution of n-hexanol (25.95 g) in dichloromethane (400 ml) was slowly added to solution of N,N-carbonyldiimidazole (48.08 g) in dichloromethane (100 ml) and stirred for 21h hour at 25-35℃.Water was added to the reaction mixture. Both the dichloromethane layer and aqueous layer were separated and the dichloromethane
layer was distilled under reduced pressure to provide the title compound.Yield: 60 g 向25.95 g正己醇400 ml二氯甲烷的溶液中缓慢滴加48.08 g N,N羰基二咪唑+100 ml二氯甲烷溶液,控温25-35℃,搅拌反应21h。反应完成后,加入纯水,分离有机相,有机相减压浓缩二氯甲烷得产品60g。
步骤3操作程序:
1-methyl-2-[N-[4-amidinophenyl]aminomethyl]benzimidazol-5-yl-carboxylicaci d-N-(2-pyridyl)-N-(2-ethoxycarbonylethyl)amide oxalate compound of formula-6a (100 g) was added to acetonitrile (1200 ml) and water (800ml) at 25-35℃. and then cooled to 12-18℃. Potassium carbonate (117 g) was added to the reaction mixture and stirred for 15 minutes at 12-18℃. A solution of hexyl 1H-imidazole-l-carboxylate compound of formula-4 (60 g) in acetonitrile (150 ml) was slowly added to the reaction mixture over a period of 25 minutes at 12-18℃. and stirred for 14 hours at 15-20℃. After completion of the reaction, water was added to the reaction mixture and stirred for 30 minutes. Filtered the solid, washed with acetonitrile followed by aqueous acetonitrile and then dried to get title compound. Dichloromethane was added to the obtained compound and stirred for 15 minutes. Water was added to the reaction mixture and stirred for 20 minutes at 25-35℃. Both the organic and aqueous layers were separated, and the dichloromethane layer was washed with water followed by sodium chloride and then distilled off completely under reduced pressure. Acetone (600 ml) was added to the obtained residue and stirred for 45 minutes at 25-35℃. to obtain a clear solution. Water (500 ml) was added to the obtained solution and stirred for 45 minutes at 25-35℃. to get the solid. Filtered the solid, washed with water and finally with methyl tertiary butyl ether and then dried to get the pure title compound. Further the obtained solid, recrystallized from ethylacetate and ethanol.
控温25-35℃,在1-甲基-2-[N-(4-脒基苯基)-氨基甲基]-苯并咪唑-5-基-羧酸-N-(2-吡啶基)-N-(2- 乙氧基羟基乙基)-酰胺草酸盐中加入乙腈1200 ml,纯水800ml。降温至12-18℃,加入碳酸钾117g,搅拌15min。控温12-18℃,在25min 内缓慢加入60g 1,1'-羰基-正己氧基-咪唑,150ml乙腈溶液,搅拌反应14h。反应完成后,加入纯水析晶搅拌30min。过滤,滤饼分别用乙腈和乙腈水溶液洗
涤,干燥。二氯甲烷溶解上步所得化合物,搅拌15min,加入纯水搅拌,静置,分层。有机相用饱和氯化钠溶液洗涤,真空浓缩干溶剂。加入丙酮600ml,控温25-35℃搅拌45min至溶液溶清,加入纯水500ml, 控温25-35℃搅拌45min,过滤,分别用纯水,甲基叔丁基醚洗涤滤饼,真空干燥得产品。
步骤4操作程序:
A solution of Dabigatran etexilate compound of formula-l (100 g) in ethyl acetate (600 ml) was heated to 40℃. and stirred for 45 minutes at 40℃Filtered the reaction mixture through hyflow bed and cooled to 25-30°C. Ethanol (60 ml) was added to the filtrate at 25-35℃. A solution of methane sulfonic acid (15 g) in ethylacetate (1000 ml) was slowly added to the above reaction mixture over a period of 2 hours at 25-35℃. and stirred for 6 hours at the same temperature. Filtered the obtained solid, washed with ethyl acetate and then dried to get the title compound.
在100g达比加群酯中加入600ml乙酸乙酯,升温至40℃,搅拌40min。通过硅藻土过滤。控温25-35℃,向滤液中加入乙醇60ml,在2h内滴加15g甲磺酸的1000ml乙酸乙酯溶液,滴加完毕后搅拌6h.过滤,滤饼用乙酸乙酯洗涤,真空干燥。
1.5、合成路线五:
N C2H5OOC NHCH3 NH2
N Cl Cl
O O
N
C2H5OOC
N
O
N
N Cl
NH2
HN
H2N
NH2
H2N
N
O
C6H13O
N
COOC2H5
N
O
N
N
HN
N
H2N
C6H13O
O
Mol. Wt.: 627.73
Mol. Wt.: 170.98
Mol. Wt.: 365.41
Mol. Wt.: 342.39
N
COOC2H5
N
O
N
N
HN
N
H2N
O
O.CH3SO3H
C35H45N7O8S
Mol. Wt.: 723.84
S OH
O
O
H3C
4
2
3
DBJQZ-0
DBJQZ-2
.2HCl
Mol. Wt.: 208.09
.HCl
DBJQZ-1
步骤1操作程序:
Compound 4 (28.0 g) is suspended in (120 mL) at 20° C. Then a mixture of ethyl acetate (50 mL) and chloroacetic acid anhydride 5b-I' (14.5g) is slowly added at 20℃. and then heated to 65℃. After 2h stirring potassium carbonate (15.0 g) is added at 40℃. and filtered after 45 min. The filter residue is washed with ethyl acetate (8.0 mL). The filtrate is evaporated down in vacuo and precipitated at 45℃. with MTBE (150 mL). It is cooled to -2℃. and filtered. The product is washed with a mixture of ethyl acetate and tert-butylmethylether (MTBE) (50 mL). The filter cake is dried in vacuo and 29.6 g product 6 (90.3% of theoretical) are obtained.
控温20℃,向28g 3-[(3-氨基-4-甲基氨基苯甲酰)吡啶-2-基氨基]丙酸乙酯中加入乙酸乙酯120ml,缓慢滴加50ml乙酸乙酯加14.5g氯乙酰酐。升温至65℃搅拌2h。降温至40℃,加入碳酸钾15g,搅拌40min。过滤,滤饼用乙酸乙酯8ml洗涤。控温45℃,真空浓缩干溶剂,加入甲基叔丁基醚150ml,降温至-2℃,过滤,滤饼用乙酸乙酯与甲基叔丁基醚的混合溶液50ml洗涤。真空干燥产品,得产品29.6g,
收率90.3%。
步骤2操作程序:
Aminobenzamidine*2HCl (21.2 g) is dissolved in acetone (150 ml), regulated to a temperature of 20℃. and sodium hydroxide solution (80 ml, 4M) is added dropwise. At 20℃. n-hexylchloroformate (16.5 g) is metered in. After rinsing with acetone (20 mL) the mixture is stirred for a further 15 min at 5-10℃. Then the phases are separated. The organic phase is evaporated down in vacuo, diluted with butyl acetate (150 mL) and the phases are separated again. The mixture is once more extracted with water (40mL) and combined with hydrochloric acid (9,84 mL, 32%). The residual water is distilled off using the water separator and then evaporated down. The suspension is mixed at 45℃with acetone (150 mL), cooled to 20℃. and suction filtered. It is washed with a mixture of butyl acetate and acetone (100 mL). The filter cake is dried in vacuo and 29.2 g of product 3 are obtained (97.2% of theoretical).
向21.2g氨基苯甲脒盐酸盐中加入150ml丙酮,调节温度为20℃,加入4M 的氢氧化钠溶液80ml。加入16.5g氯甲酸正己酯,并用20ml丙酮清洗。降温至5-10℃.搅拌15min,静置分层,有机层减压浓缩溶剂,加入乙酸丁酯150ml,搅拌静置分层。有机层用纯水40ml、32%盐酸9.84ml萃取。残余的水用分水器分离,真空浓缩干溶剂。控温45℃,加入丙酮150ml,冷却至20℃,抽滤,用乙酸丁酯和丙酮的混合溶剂100ml洗涤,真空干燥,得产品29.2g,收率97.2%。
步骤3操作程序:
Compound 3 (7.7 g) is placed in butyl acetate (65mL), sodium hydroxide solution (25 mL, 45%) and water (25mL) and heated to 50℃. Then the phases are separated and the organic phases are extracted again with water (30 mL).The organic phase is combined with sodium iodide (1.54 g),sodium hydrogen carbonate (4.00 g), tetrabutylammonium iodide (0.75 g), compound 6 (10.0 g), cyclohexane (65 mL) and water (30 mL) and stirred for 2 h at 40℃. Then the cyclohexane is distilled off in vacuo, butyl acetate (95 mL) is added and the mixture is stirred for 2 h at 70℃. Then the phases are separated and the organic phase is extracted twice with water (10 mL). The organic phase is evaporated down in vacuo, the solution is cooled to 0℃. and filtered. The product is washed with butyl acetate (30 mL). The filter cake is dried in
vacuo and 13.8 g product 7 are obtained (87.8% oftheoretical).
向第2步所得产物7.7g中加入65ml乙酸丁酯,45%氢氧化钠溶液25ml,纯水25ml,升温至50℃。静置分层,分离有机相,有机相用纯水30ml洗涤。有机相加入碘化钠1.54 g、碳酸氢钠4.0 g、四丁基碘化铵0.75g 、第1步所得化合物10.0 g、环己烷65ml、纯水30ml,升温至40℃,搅拌2h。减压浓缩掉环己烷,加入乙酸丁酯95ml,升温至70℃,搅拌2h。静置分层,有机相用纯水20ml洗涤2次。真空浓缩干溶剂,冷却至0℃,过滤,用乙酸丁酯30ml洗涤,真空干燥得产品13.8g,收率87.8%。
步骤4操作程序:
Compound 7 (20 g) is suspended in acetone (238mL) at ambient temperature and refluxed. The solution is filtered clear and rinsed with acetone (20 mL). The filtrate is cooled to 33℃. and a solution of methanesulphonic acid (3.0g) in acetone (34 mL) cooled to 0℃. is metered in and the mixture is rinsed with acetone (5.0 mL). Then it is cooled to 20℃. and filtered. The product is washed with acetone (54mL). The filter cake is dried in vacuo and 22.2 g product 8 are obtained (96.3% of theoretical).
将第3步所得产物20g悬浮在238ml丙酮中,升温至回流搅拌,趁热过滤,用丙酮20ml洗涤。降温至33℃,滴加0℃的甲磺酸(3.0g)丙酮(34 mL)溶液,用5ml丙酮冲洗加液滤斗。冷却至20℃,过滤,滤饼用丙酮54ml洗涤,真空干燥的产品22.2g,收率96.3%。
物料
211915-84-3 530-62-1 1307233-93-7
4-氨基苯甲脒二盐酸盐
2498-50-2氯甲酸正己酯
6092-54-2
氯乙酸酐
541-88-8
872728-82-0
达比加群酯用于非瓣膜病心房颤动患者卒中预防的临床应用建议 2004年的流行病学调查显示,我国心房颤动(房颤)患病率约为0.77%。按人口推算,我国30岁以上房颤患者数为420万。随着人口的老龄化,房颤的患病率将持续增长。缺血性脑卒中(卒中)是房颤最为严重的并发症,与无房颤患者相比,房颤患者的卒中风险升高约5倍,且房颤相关卒中致残和致死率更高。因此,预防卒中是房颤综合管理的 重要策略。 抗凝治疗是房颤患者卒中预防的基石。华法林是循证证据最充分、使用最普遍的口服抗凝药物,但由于剂量个体差异大、药物-药物/食物相互作用常见,需频繁监测,加上医生对华法林所致出血的过度担心,影响了其在临床实践中的广泛应用。临床上亟需疗效可靠、安全性更优的新型口服抗凝药物。 2010-2011年直接凝血酶抑制剂达比加群在欧美、2013年在我国获得批准用于预防非瓣膜性房颤患者卒中。本建议针对达比加群酯临床应用中的常见问题,将目前的国内外临床应用经验进行总结,澄清一些临床使用误区,为临床医生提供详实、准确、实用的达比加群酯临床指导方案。 一、达比加群酯的作用机制和药代动力学 达比加群酯为前体药物,口服后经肝脏被酯酶转化为活性代谢产物,达比加群为直接凝血酶抑制剂,以浓度依赖方式特异性阻断凝血酶(IIa因子)活性,其不仅可与游离型 IIa因子结合,还可与血栓结合型IIa因子结合。阻断IIa因子阻断了凝血瀑布网络的最后 步骤。
达比加群的亲水性极性分子结构使其难以通过肠道被吸收,在其分子结构上添加一个亲脂性侧后成为达比加群酯,使胃肠道吸收率升高1倍。达比加群酯口服后,迅速在胃和小肠吸收,并通过酯酶催化水解作用变成活性产物——达比加群。这种蛋白水解过程不受细胞色素P450同功酶或其他氧化还原酶影响。另外,体外研究发现,达比加群也不抑制细胞色素P450同功酶活性。因此,达比加群酯与其他药物很少出现相互作用。 口服给药后,达比加群的绝对生物利用度为6%~7%。吸收迅速,2小时内达最大血药浓度(C max),与食物同时服用可使C max延后2小时。达比加群的平均终末半衰期在 健康老年人中约11小时。多次给药后终末半衰期约12~14小时, 2~3天后达稳态。肾功能不全时半衰期延长。达比加群具有中度的组织分布,分布容积60~70L。静脉给药后,主要以原形经尿液排泄(85%),清除率与肾小球滤过率相应,约100ml/min,粪便排 泄占给药剂量的6%。达比加群在患者体内迅速起效,起效时间为0.5~2小时;随着达 比加群的药物浓度增加,活化部分凝血活酶时间(aPTT)延长。 二、达比加群酯的临床研究 达比加群酯用于非瓣膜性房颤患者的疗效和安全性在长期抗凝治疗随机评估研究(Randomized Evaluation of Long-Term Anticoagulation Therapy,RE-LY)中得到验证[9-11]。RE-LY试验是一项随机临床非劣效性研究,在全球44个国家的951个中心入 选伴有至少一项卒中风险的房颤患者18113例,随机分三组,两组患者以盲法接受固定 剂量达比加群酯(110 mg或150 mg,每日2次)治疗,一组以非盲法接受调整剂量的华法林治疗,主要评价三组患者的卒中或全身性栓塞以及大出血的发生率。经过中位数 2.0年的随访,结果发现,达比加群酯150mg组卒中/全身性栓塞的年化发生率较华法林组显著降低35%(1.11%对 1.71%,优效检验P<0.001),大出血风险相当( 3.32% 对 3.57%,P=0.32);而达比加群酯110mg组的卒中/全身性栓塞的年化发生率与华法
达比加群酯临床应用建 议 Coca-cola standardization office【ZZ5AB-ZZSYT-ZZ2C-ZZ682T-ZZT18】
达比加群酯用于非瓣膜病心房颤动患者卒中预防的临床应用建议 2004年的流行病学调查显示,我国心房颤动(房颤)患病率约为%。按人口推算,我国30岁以上房颤患者数为420万。随着人口的老龄化,房颤的患病率将持续增长。缺血性脑卒中(卒中)是房颤最为严重的并发症,与无房颤患者相比,房颤患者的卒中风险升高约5倍,且房颤相关卒中致残和致死率更高。因此,预防卒中是房颤综合管理的重要策略。 抗凝治疗是房颤患者卒中预防的基石。华法林是循证证据最充分、使用最普遍的口服抗凝药物,但由于剂量个体差异大、药物-药物/食物相互作用常见,需频繁监测,加上医生对华法林所致出血的过度担心,影响了其在临床实践中的广泛应用。临床上亟需疗效可靠、安全性更优的新型口服抗凝药物。 2010-2011年直接凝血酶抑制剂达比加群在欧美、2013年在我国获得批准用于预防非瓣膜性房颤患者卒中。本建议针对达比加群酯临床应用中的常见问题,将目前的国内外临床应用经验进行总结,澄清一些临床使用误区,为临床医生提供详实、准确、实用的达比加群酯临床指导方案。一、达比加群酯的作用机制和药代动力学 达比加群酯为前体药物,口服后经肝脏被酯酶转化为活性代谢产物,达比加群为直接凝血酶抑制剂,以浓度依赖方式特异性阻断凝血酶(IIa因
子)活性,其不仅可与游离型IIa 因子结合,还可与血栓结合型IIa 因子结合。阻断IIa 因子阻断了凝血瀑布网络的最后步骤。 达比加群的亲水性极性分子结构使其难以通过肠道被吸收,在其分子结构上添加一个亲脂性侧后成为达比加群酯,使胃肠道吸收率升高1倍。达比加群酯口服后,迅速在胃和小肠吸收,并通过酯酶催化水解作用变成活性产物——达比加群。这种蛋白水解过程不受细胞色素P450同功酶或其他氧化还原酶影响。另外,体外研究发现,达比加群也不抑制细胞色素P450同功酶活性。因此,达比加群酯与其他药物很少出现相互作用。 口服给药后,达比加群的绝对生物利用度为6%~7%。吸收迅速,2小时内达最大血药浓度(C max ),与食物同时服用可使C max 延后2小时。达比加 群的平均终末半衰期在健康老年人中约11小时。多次给药后终末半衰期约12~14小时, 2~3天后达稳态。肾功能不全时半衰期延长。达比加群具有中度的组织分布,分布容积60~70L 。静脉给药后,主要以原形经尿液排泄(85%),清除率与肾小球滤过率相应,约100ml/min ,粪便排泄占给药剂量的6%。 达比加群在患者体内迅速起效,起效时间为~2小时;随着达比加群的药物浓度增加,活化部分凝血活酶时间(aPTT )延长。 二、 达比加群酯的临床研究
如对你有帮助,请购买下载打赏,谢谢! 达比加群酯用于非瓣膜病心房颤动患者卒中预防的临床应用建议 2004年的流行病学调查显示,我国心房颤动(房颤)患病率约为0.77%。按人口推算,我国30岁以上房颤患者数为420万。随着人口的老龄化,房颤的患病率将持续增长。缺血性脑卒中(卒中)是房颤最为严重的并发症,与无房颤患者相比,房颤患者的卒中风险升高约5倍,且房颤相关卒中致残和致死率更高。因此,预防卒中是房颤综合管理的重要策略。 抗凝治疗是房颤患者卒中预防的基石。华法林是循证证据最充分、使用最普遍的口服抗凝药物,但由于剂量个体差异大、药物-药物/食物相互作用常见,需频繁监测,加上医生对华法林所致出血的过度担心,影响了其在临床实践中的广泛应用。临床上亟需疗效可靠、安全性更优的新型口服抗凝药物。 2010-2011年直接凝血酶抑制剂达比加群在欧美、2013年在我国获得批准用于预防非瓣膜性房颤患者卒中。本建议针对达比加群酯临床应用中的常见问题,将目前的国内外临床应用经验进行总结,澄清一些临床使用误区,为临床医生提供详实、准确、实用的达比加群酯临床指导方案。 一、达比加群酯的作用机制和药代动力学 达比加群酯为前体药物,口服后经肝脏被酯酶转化为活性代谢产物,达比加群为直接凝血酶抑制剂,以浓度依赖方式特异性阻断凝血酶(IIa因子)活性,其不仅可与游离型IIa因子结合,还可与血栓结合型IIa因子结合。阻断IIa因子阻断了凝血瀑布网络的最后步骤。 达比加群的亲水性极性分子结构使其难以通过肠道被吸收,在其分子结构上添加一个亲脂性侧后成为达比加群酯,使胃肠道吸收率升高1倍。达比加群酯口服后,迅速在胃和小肠吸收,并通过酯酶催化水解作用变成活性产物——达比加群。这种蛋白水解过程不受细胞色素P450同功酶或其他氧化还原酶影响。另外,体外研究发现,达比加群也不抑制细胞色素P450同功酶活性。因此,达比加群酯与其他药物很少出现相互作用。 口服给药后,达比加群的绝对生物利用度为6%~7%。吸收迅速,2小时内达最大血药浓度(C max),与食物同时服用可使C max延后2小时。达比加群的平均终末半衰期在健康老年人中约11小时。多次给药后终末半衰期约12~14小时, 2~3天后达稳态。肾功能不全时半衰期延长。达比加群具有中度的组织分布,分布容积60~70L。静脉给药后,主
达比加群酯胶囊说明书 达比加群酯胶囊(泰毕全)用于预防心节律异常(心房颤动)患者中风和血栓的发生。下面是学习啦小编整理的达比加群酯胶囊说明书,欢迎阅读。 达比加群酯胶囊商品介绍通用名:达比加群酯胶囊生产厂家: Boehringer Ingelheim International GmbH. 批准文号:国药准字J20130064 药品规格:110mg*10粒 药品价格:¥198元 达比加群酯胶囊说明书【通用名称】达比加群酯胶囊 【商品名称】达比加群酯胶囊(泰毕全) 【英文名称】DabigatranEtexilateCapsules(Pradaxa) 【拼音全码】DaBiJiaQunZhiJiaoNang(TaiBiQuan) 【主要成份】甲磺酸达比加群酯。 化学名:3-[[[2-[[[4-[[[(己氧基)羰基]氨基]亚氨甲基]苯基]氨基]甲基]-1-甲基-1H-苯并咪唑-5-基]羰基](吡啶-2-基)氨基]丙酸乙酯甲磺酸盐 分子式:C34H41N7O5CH4O3S 分子量:723.86 【性状】达比加群酯胶囊(泰毕全)为胶囊剂,内容物为
黄色颗粒。 【适应症/功能主治】用于预防心节律异常(心房颤动)患者中风和血栓的发生。 【规格型号】110mg*10s 【用法用量】用水送服,餐食或餐后服用均可。请勿打开胶囊。成人的推荐剂量为每日口服300mg,即每次1粒150mg的胶囊,每日两次,应维持终生治疗。 【不良反应】1.在关键部位或器官发生症状性出血:眼内、颅内、椎管内或伴有骨筋膜室综合征的肌肉内出血、腹膜后出血、关节内出血或心包出血。2.满足以下一项或一项以上标准的大出血被称为危及生命的出血:致死性出血、症状性颅内出血;伴有血红蛋白至少下降50g/L的出血;需要输血或血细胞至少达4个单位的出血,伴有低血压而需静脉使用升压药的出血;必须外科手术治疗的出血。3.与接受华法林治疗者相比,随机接受达比加群酯每次110mg、每日两次和每次150mg.每日两次的患者,总体出血、危及生命的出血和颅内出血风险呈显著下降(p 【禁忌】1.已知对活性成分或达比加群酯胶囊(泰毕全)任一辅料过敏者。2.重度肾功能不全(CrCl 【注意事项】1.房颤相关性卒中和SEE预防的临床试验中排除了肝酶增高>2ULN(正常值上限)的患者。对这一患者亚组无治疗经验,所以不推荐该人群使用达比加群酯胶囊(泰毕全)。2.与其他所有抗凝药物一样,出血
PRADAXA?(甲磺酸达比加群酯dabigatran etexilate mesylate)使用说明书2012年12月版 处方资料重点 这些重点不包括安全和有效使用PRADAXA所需所有资料。请参阅下文为PRADAXA的完整处方资料 PRADAXA? (甲磺酸达比加群酯[dabigatran etexilate mesylate])胶囊为口服使用 美国初始批准:2010 最近重大变化(红色为修改部分) 剂量和给药方法(2.2,2.4,2.6) 1/2012 禁忌证(4) 12/2012 警告和注意事项(5.3,5.4) 1/2012 警告和注意事项(5.1) 11/2012 警告和注意事项(5.2) 12/2012 适应证和用途 PRADAXA是一种直接凝血酶抑制剂适用于在有非瓣膜性房颤患者中减低卒中和全身性栓塞风险(1) 剂量和给药方法 (1)对患者有CrCl >30 mL/min:150 mg口服,每天2次 (2.1) (2)对患者有CrCl 15-30 mL/min:75 mg口服,每天2次 (2.1) (3)治疗期间临床上指示时评估肾功能和从而调整治疗(2.2) (4)指导患者不要咀嚼,弄破,或打开胶囊 (2.3) (5)对转换至或来其他口服或非肠道抗凝建议的评述 (2.4,2.5) (6)侵入性或手术前当可能暂时终止PRADAXA,然后及时再开始(2.6)
剂型和规格 胶囊:75 mg和150 mg (3) 禁忌证 (1)活动性病理性出血 (4) (2)对PRADAXA严重超敏性反应史(4) (3)机械性人工心脏瓣膜 (4) 警告和注意事项 (1)出血风险:PRADAXA可引起严重和,有时,致命性出血。及时评价失血的体征和症状。 (5.1) (2)生物人工心脏瓣膜:建议不要用PRADAXA(5.2) (3)暂时终止:避免治疗失误缩小卒中风险(5.3) (4)P-gp 诱导剂和抑制剂:对达比加群暴露影响 (5.4) 不良反应 最常见不良反应(>15%)是胃炎样症状和出血(6.1) 为报告怀疑不良反应,联系Boehringer Ingelheim Pharmaceuticals,Inc. 电话(800) 542-6257或(800) 459-9906 TTY或FDA电话1-800-FDA-1088或https://www.doczj.com/doc/c95012194.html,/medwatch. 药物相互作用 (1)P-gp诱导剂利福平[利福平]:避免与PRADAXA共同给药 (5.4) (2)P-gp抑制剂决奈达隆[dronedarone]和全身性酮康唑[ketoconazole]在患者有中度肾受损(CrCl 30-50 mL/min):考虑减低PRADAXA剂量至75 mg每天2次 (7) (3)P-gp抑制剂在有严重肾受损患者(CrCl <30 mL/min):建议不使用PRADAXA (7) 特殊人群中使用 老年人使用:随年龄出血风险增加 (8.5) 完整处方资料 1 适应证和用途 PRADAXA适用于在有非瓣膜性房颤患者中减低卒中和全身性栓塞的风险。 2 剂量和给药方法
达比加群酯的合成方法研究进展 本文从网络收集而来,上传到平台为了帮到更多的人,如果您需要使用本文档,请点击下载按钮下载本文档(有偿下载),另外祝您生活愉快,工作顺利,万事如意! 抗凝血药是一类通过影响凝血过程某些凝血因子,阻止血液凝固的药物,主要用于防治血栓栓塞性疾病及其并发症。达比加群酯是由德国勃林格殷格翰公司研发的一种新型非肤类直接凝血酶抑制剂,于2008年在欧盟获准用于全髓或全膝关节置换手术后静脉血栓的预防;2010年获得美国FDA批准上市,用于减少非瓣膜性心房颤动患者脑卒中及全身血栓栓塞的预防[l];2013年获我国食品药品监督管理局颁发的进口药品注册证。 达比加群酯是一种前药,本身没有活性,口服经胃肠吸收后,在肝脏中迅速被酯酶水解并进一步转化为有活性的达比加群(Dahigatran),水解过程如下所示。后者通过结合于凝血酶的纤维蛋白特异位点,阻止纤维蛋白原裂解为纤维蛋白,从而阻断凝血瀑布网络的最后步骤及血栓形成。 达比加群酯具有可口服、强效、无需特殊用药监测、药物相互作用少等优点,其合成受到国内外研究人员的广泛关注,己报道多条合成路线。本文综述了
达比加群酯的合成方法。 1达比加群酯的合成方法 1. 1直接合成法 Haul等最先合成了达比加群酯,并发现它的优异抗凝作用。其合成路线是以肖基4-氯苯甲酸(2)为起始原料,首先与甲胺水溶液缩合得4-甲氨基-不肖基苯甲酸再经氯化亚飒。 达比加群酯的合成路线氯代,然后与氨基)丙酸乙酯缩合得3-(4-(甲基氨基)一不肖基W-(毗咙2长基)苯甲酞氨基)丙酸乙酯(6)。化合物6经把碳催化氢化得3- [(3氰基4一甲基氨基苯甲酞)毗咙2基氨基]丙酸乙酯(7),后者再经N-( 4债氯基苯基)甘氨酸(8)酞胺化,然后闭环得到氰基化合物9,化合物9相继用氯化氢的饱和乙醇溶液和碳酸铰的乙醇溶液处理,发生Pinned反应制得眯类化合物10,然后与氯甲酸正己酯(11)缩合制得达比加群酯(1)。该方法的优点是操作简便、条件温和、成本较低,缺点是制备酞氯需使用二氯亚飒,成眯反应需要通入干燥氯化氢气体,两者具有强腐蚀性和刺激性,后处理麻烦且污染环境。此后的一些合成方法基本上都是在此基础上进行改进而得到的。 1. 2成盐法
达比加群酯研发经历总结 摘要 达比加群酯,美国FDA和欧盟分别于2010年l 0月19日和2008年3月18日批准Boellringer Ingelllein制药公司的达比加群醋胶囊用于心房颤动患者预防中风和血栓生成。分子式为C34H41N7O5,相对分子质景为723.86。达比加群酯是最前沿的新一代口服抗凝药物直接凝血酶抑制剂(DTIs),针对急性和慢性血栓栓塞性疾病的预防及治疗这一急需满足的临床需求。 Dabigatran etexilate, American FDA and European Union respectively in 2010 L 0 on 19 March and March 18, 2008 approved Boellringer Ingelllein pharmaceuticals of dabigatran vinegar capsule for the prevention of stroke in patients with atrial fibrillation and thrombus formation. The molecular formula is C34H41N7O5, relative molecular quality 723.86. Dabigatran is a new oral anticoagulant direct thrombin inhibitor most front (DTIs), according to clinical demand for prevention of acute and chronic thromboembolic disease and treatment of the need to meet. 药物简介 达比加群酯是一种新型的合成的直接凝血酶抑制剂,是dabigatran的前体药物,属非肽类的凝血酶抑制剂。口服经胃肠吸收后,在体内转化为具有直接抗凝血活性的dabigatran。dabigatran结合于凝血酶的纤维蛋白特异结合位点,阻止纤维蛋白原裂解为纤维蛋白,从而阻断了凝血瀑布网络的最后步骤及血栓形成。dabigatran可以从纤维蛋白一凝血酶结合体上解离,发挥可逆的抗凝作用。 Dabigatran is a new synthetic direct thrombin inhibitor, is a prodrug of dabigatran, is a thrombin inhibitor non-peptide. Oral absorbed by the gastrointestinal, converted in the body to have direct anticoagulant activity of dabigatran. Fibrin specific dabigatran binding to thrombin binding sites, preventing fibrinogen lysis fibrin, thereby blocking the last step and thrombus coagulation cascade network formation. Dabigatran can be from a combination of fibrinogen thrombin dissociation, play the anticoagulant effect of reversible.
达比加群酯合成路线综述 一、 合成路线 1.1、 合成路线一: N COOC 2H 5 H 3CHN H 2N N O NC NHCH 2COOH ,HOBT,EDCl N COOC 2H 5 N O N N HN NC N COOC 2H 5 N O N N HN HN H 2N O O 1 3 N COOC 2H 5 N O N N HN N H 2N O O Mol. Wt.: 627.73 Mol. Wt.: 164.63 Mol. Wt.: 499.56 Mol. Wt.: 482.53 Mol. Wt.: 176.17 Mol. Wt.: 342.39 1) HCl/EtOH 2) (NH 4)2CO 3/EtOH 步骤1操作程序: 将6.17 g (0.035 mol)N-(4-氰基苯基)甘氨酸及5.68 g (0.035 mol),N,N'-羰基 二咪唑在300 毫升四氢呋喃中加热回流30分钟,然后加入10.6 g(0.032 mol) 3-氨基-4-甲基氨基-苯甲酸-N-(2-吡啶基)N-(2-乙氧基羰基乙基)-酰胺,将该混合物加热回流5小时。然后真空蒸馏去除溶剂,将残余物溶于150 ml 冰醋酸内,加热回流一小时。然后真空蒸馏去除冰醋酸,将残余物溶于约300 ml 二氯甲烷内,该溶液用约150 ml 水洗二次,然后于硫酸钠上干燥。蒸发去除溶剂后将所得粗制产物作柱色层纯化(800 g 硅胶;洗脱剂:二氯甲烷及1-2 % 乙醇)。产量8.5g ,收率57%,Rf 值0.51(二氯甲烷:乙醇=19:1)。 步骤2操作程序: 将1.2 g (2 .49 mmol) 1-甲基-2-[N-(4-氰基苯基)-氨基甲基]-苯并咪唑-5-基-羧
达比加群酯合成工艺杂质分析 一.达比加群酯结构式 二.合成路线: 1.成脒反应 2.酰化反应 3.达比加群酯甲磺酸盐合成 三.达比加群酯杂质来源分析: 通过达比加群酯合成文献及结构式推测了以下杂质的可能的产生来源:1.达比加群酯杂质ZA 该杂质可能来源是在酰化反应中,四氢呋喃-水-氢氧化钾体系下达比加群酯发生酯基水
解反应。也可能是该步中间体(2)首先发生水解反应生成达比加群酯杂质ZB,后发生酰化反应生成。 2.达比加群酯杂质ZB 该杂质可能来源是在酰化反应中,四氢呋喃-水-氢氧化钾体系下中间体(2)生酯基水解反应。 3.达比加群酯杂质ZC 该杂质可能来源是在成脒反应中产生的杂质(达比加群酯杂质ZM)在酰化反应发生反应而产生。 4.达比加群酯杂质ZD 该杂质可能来源是在酰化反应中由酰化剂引入的杂质而产生的杂质。 5.达比加群酯杂质ZE
该杂质可能来源是在酰化反应中由酰化剂引入的杂质而产生的杂质。 6.达比加群酯杂质ZF 该杂质可能来源是是制备中间体(1)的过程中原料与溶剂醋酸反应生成,如下图所示。 7.达比加群酯杂质ZG 该杂质可能来源是是制备中间体(1)的过程中原料与缩合剂CDI(羰基二咪唑)反应生成,如下图所示。 8.达比加群酯杂质ZH 该杂质可能来源是在成脒反应中产生的杂质在酰化反应发生反应而产生。
9.达比加群酯杂质ZI 该杂质为合成路线中间体(1),残留引入终产品。 10.达比加群酯杂质ZJ 该杂质为在成脒反应中产生亚胺酯中间体,残留引入终产品。 11.达比加群酯杂质ZK 该杂质可能为合成路线中与某一步与甲醇发生酯交换反应生成,残留引入终产品;或者由合成原料中的甲酯杂质引入。 12.达比加群酯杂质ZL
达比加群Pradaxa:向抗凝治疗三大误区说“不” 心房颤动(简称“房颤”)是最常见的心率失常。随着人口老龄化和慢性心脏病的影响,房颤的全球发病率与日俱增,总发病率达0.4%,且随年龄增长而不断增加,75岁以上人群达10%1。在中国,房颤的患病现状更令人堪忧,据“全球预防中风行动”专家组发布的《如何避免亚太地区卒中危机》报告,目前中国约有800万房颤患者,数量超过欧美国家的总和,成为房颤“第一大国”2。 房颤患者在发病时,心房跳动的频率极快且不规整,每分钟“颤动”高达350-600次1,轻者会感觉心慌、气短,重者则会诱发脑卒中等血栓性疾病和心力衰竭。那么,只是“心律失常”的房颤,怎么会引发脑卒中呢?专家们指出,患者在房颤时,会使流过心房的血液流速变慢,容易淤积形成血栓;形成的血栓粘在心房壁上,心房在颤动中,血栓就会脱落并随血流到心室,然后再达到全身。大多数情况下,心脏把血栓往上泵,堵塞脑血管后就会发生脑卒中,俗称“中风”。 由于脑卒中会引发高致残率和致死率,目前在全球范围内,各指南、共识,包括《2012年心房颤动管理指南更新》和《新房颤动抗凝治疗中国专家共识》都已将预防脑卒中列为房颤治疗的重要目标之一。而被证实能实现这一目标最有效的方法就是抗凝治疗。抗凝治疗的原理是通过影响血液凝固以维持血液的正常流动,使血管畅通无阻,通俗点说,就是使血液在血管里“不堵车”。但根据2010年全球性房颤注册的调查数据,我国房颤抗凝治疗率仅为10%3,“预防脑中风”显然已经成为了房颤治疗中的一句“空口号”。究其原因,主要是医生和患者对抗凝治疗存有误区。 误区一:对抗凝药存在的出血风险“诚惶诚恐”,面对房颤背后的中风风险直接举白旗投降。事实上,任何预防中风的抗凝治疗都存在出血风险。影响出血并发症的因素有很多,包括年龄、抗凝强度、抗凝时间和药物之间的相互作用等。当出现出轻微出血症状时,医生和患者不必过分恐慌,而是应该及时了解出血原因,有针对性地采取措施。但绝不能因此而拒绝接受科学、积极的抗凝治疗,因为由房颤引发的中风可能导致比轻微出血更严重百倍的临床后果。目前,传统抗凝药华法林是最常用的抗凝剂,但一项针对华法林使用者的大型观察研究结果表明,华法林的主要出血事件率在1%-3%之间4。与
“十三五”重点项目-达比加群酯甲磺酸盐项目可行性研究报告
————————————————————————————————作者:————————————————————————————————日期:
“十三五”重点项目-达比加群酯甲磺酸盐项目可行性研究报告 编制单位:北京智博睿投资咨询有限公司
本报告是针对行业投资可行性研究咨询服务的专项研究报告,此报告为个性化定制服务报告,我们将根据不同类型及不同行业的项目提出的具体要求,修订报告目录,并在此目录的基础上重新完善行业数据及分析内容,为企业项目立项、申请资金、融资提供全程指引服务。 可行性研究报告是在招商引资、投资合作、政府立项、银行贷款等领域常用的专业文档,主要对项目实施的可能性、有效性、如何实施、相关技术方案及财务效果进行具体、深入、细致的技术论证和经济评价,以求确定一个在技术上合理、经济上合算的最优方案和最佳时机而写的书面报告。 可行性研究是确定建设项目前具有决定性意义的工作,是在投资决策之前,对拟建项目进行全面技术经济分析论证的科学方法,在投
资管理中,可行性研究是指对拟建项目有关的自然、社会、经济、技术等进行调研、分析比较以及预测建成后的社会经济效益。在此基础上,综合论证项目建设的必要性,财务的盈利性,经济上的合理性,技术上的先进性和适应性以及建设条件的可能性和可行性,从而为投资决策提供科学依据。 投资可行性报告咨询服务分为政府审批核准用可行性研究报告和融资用可行性研究报告。审批核准用的可行性研究报告侧重关注项目的社会经济效益和影响;融资用报告侧重关注项目在经济上是否可行。具体概括为:政府立项审批,产业扶持,银行贷款,融资投资、投资建设、境外投资、上市融资、中外合作,股份合作、组建公司、征用土地、申请高新技术企业等各类可行性报告。 报告通过对项目的市场需求、资源供应、建设规模、工艺路线、设备选型、环境影响、资金筹措、盈利能力等方面的研究调查,在行业专家研究经验的基础上对项目经济效益及社会效益进行科学预测,从而为客户提供全面的、客观的、可靠的项目投资价值评估及项目建设进程等咨询意见。 报告用途:发改委立项、政府申请资金、申请土地、银行贷款、境内外融资等 关联报告: 达比加群酯甲磺酸盐项目建议书 达比加群酯甲磺酸盐项目申请报告 达比加群酯甲磺酸盐资金申请报告
中文名称英文名称CAS 规格用途结构式 达比加群酯Pradaxa 872728-81-910mg-25mg-50mg-100mg 项目报批 纯度高于98.89% 去乙基达比加群酯Desethyl Pradaxa 212321-78-310mg-25mg-50mg-100mg 项目报批 纯度高于98.89% 武汉斯坦德供应各种杂质对照品:泊沙康唑杂质、替卡格雷杂质、索拉非尼杂质、索拉菲尼相关杂质、去氧肾上腺素杂质、维生素BI杂质、马来酸氯苯那敏杂质、瑞格列奈杂质等;并提供COA、NMR、HPLC、MS等图谱。详情请点用户名。 专注各种杂质对照品 代理中检所/EP/BP/USP/LGC/TRC/DR/TLC/MC/SIGMA/BACHEM/STD等品牌达比加群酯Pradaxa杂质整理列表 1. 药典标准品: 代理销售 CP( 中检所标准品 )/EP( 欧洲药典 )/BP (英国药典)/USP (美国药典) /JP (日本药典) / WHO (世界卫生组织药物标准品)。产品种类齐全,到货迅速。 2. 药物杂质对照品: 专业提供新药申报所需各种药物杂质对照品,药物中间体及代谢产物。主要代理品牌: LGC,TRC,TLC, MC等。 3. 原研参比制剂: 我们提供非临床研究用的 ICH 成员国参比制剂 / 海外上市品 /对照药品,作为科学研究用样品(国外对照品)。作为临床试验用对照品(制剂)则需要向国家食品药品监督管理局申请一次性药品进口的许可,是一个复杂的过程。我们有能力提供高效可靠的世界范围的国外对照品采购服务,代为向主管部门办理申请的程序。我们的服务快速、准确,严格遵守道德规范。 4. 超精细化学品及定制产品: 斯坦德化工技术有限公司与全球众多科研实验室建立了良好的合作关系,根据客户需要定制生产药物杂质,中间体,代谢物等,快速满足各科研单位的需求。