
原核表达步骤
原核表达先要将基因克隆到原核表达载体上,然后通过转化到
JM109或BL21等菌株中,诱导表达蛋白,然后进行蛋白纯化。本实验方案的前提是,目的基因已克隆到载体,并已转进入JM109菌株中。
1.鉴定目的蛋白是否在大肠杆菌JM109或BL21中大量表达
(1)制样
1 . 挑取经过双酶切鉴定的单克隆菌落于700ul LB培养基,加入0.7ul Amp(100mg/mL),37o C200r/min摇床培养,过夜活化。
2. 以1:50比例(200ul),将活化的过夜培养物加入10mL LB液体培养基中,加入10uLAmp(100mg/ml),37o C200r/min摇床扩大培养2h-3h,期间取样监控菌液的OD值,控制菌液OD600在0.6-1.0之间,以使大肠杆菌处于最适合表达外源蛋白的生长状态。(一般3h时,菌液浓度及达到标准,但是不同的基因对菌的影响不同,所以第一次实验时需要确定这个最佳时间)
3. 从10ml扩大培养物中取3ml菌液作为不加IPTG的空白对照(CK),其余7ml菌液加入7ul IPTG(储存浓度为0.5mol/l),使IPTG 终浓度达到0.5mmol/l。以200r/min的转速,37o C摇床培养3h。
4. 以5000r/min离心2min收集菌体,倾倒上清,每个离心管收集3ml培养物。
5. 加入1ml dH2O,将管底沉淀用振荡器打散以充分洗涤,8000r/min 离心2min,倾倒上清。
6. 重复步骤5。将离心管中的水倒干净。
(二)菌落SDS-PAGE
1. 在收集的菌体中加入200ul 1×SDS PAGE loading buffer(可根据沉淀的量增加或减少loading buffer的量,一般200ul比较合适)。用漩涡器剧烈震荡,确保将管底沉淀震散。
2. 将样品于100℃恒温加热器上开盖加热10min(Marker也要加热)。样品凉后,12000r/min离心3min,取每管的上清点样。上样量一般30ul—40ul,marker 20ul。
(3)SDS-PAGE分析
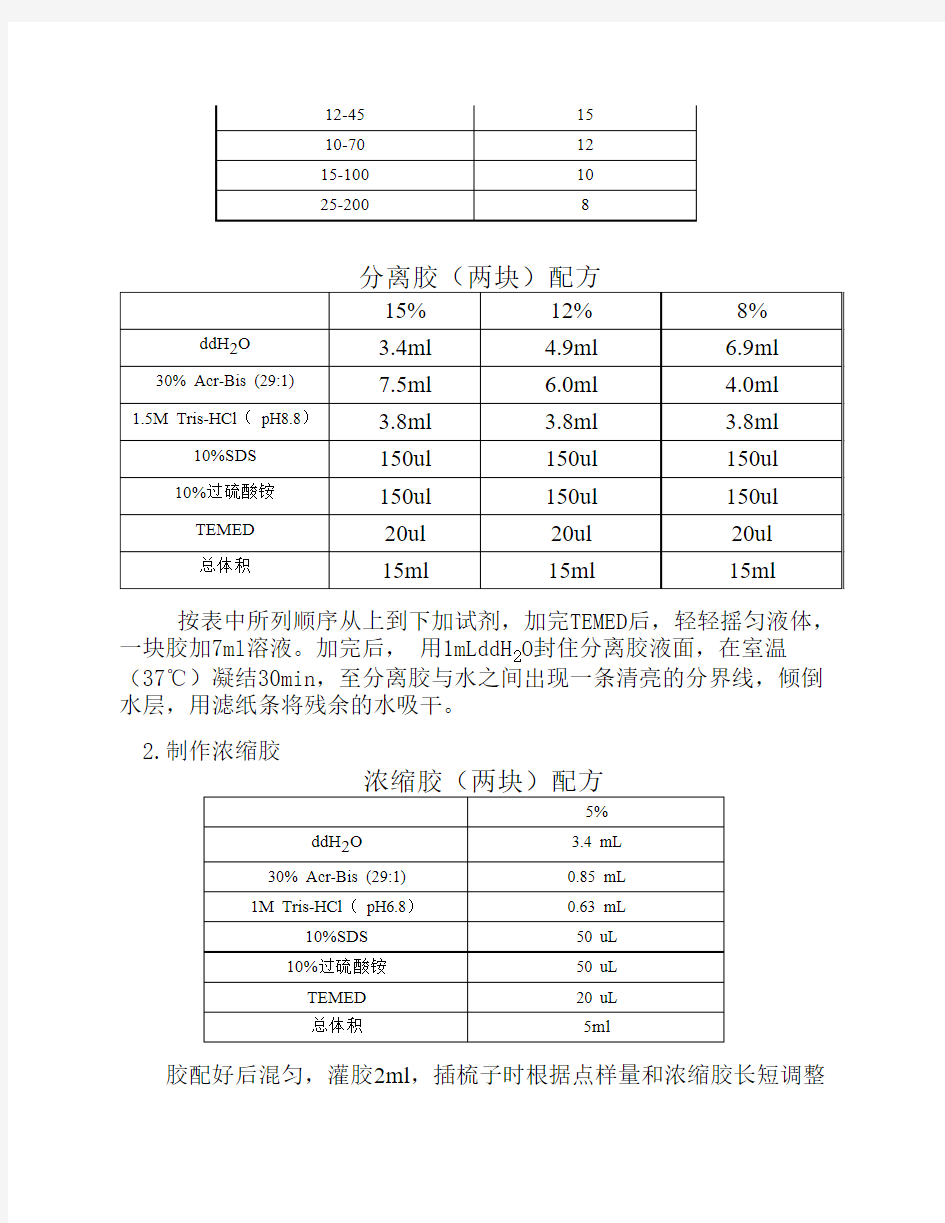
1. 根据目的片段的大小,制作不同浓度的分离胶
蛋白分子量 (kDa)凝胶浓度 (%)
4-4020
12-4515
10-7012
15-10010
25-2008
分离胶(两块)配方
15%12%8% ddH2O 3.4ml 4.9ml 6.9ml 30% Acr-Bis (29:1)7.5ml 6.0ml 4.0ml
1.5M Tris-HCl( pH8.8) 3.8ml 3.8ml 3.8ml
10%SDS150ul150ul150ul
10%过硫酸铵150ul150ul150ul
TEMED20ul20ul20ul
总体积15ml15ml15ml
按表中所列顺序从上到下加试剂,加完TEMED后,轻轻摇匀液体,一块胶加7ml溶液。加完后, 用1mLddH2O封住分离胶液面,在室温(37℃)凝结30min,至分离胶与水之间出现一条清亮的分界线,倾倒水层,用滤纸条将残余的水吸干。
2.制作浓缩胶
浓缩胶(两块)配方
5%
ddH2O 3.4 mL
30% Acr-Bis (29:1)0.85 mL
1M Tris-HCl( pH6.8)0.63 mL
10%SDS50 uL
10%过硫酸铵50 uL
TEMED20 uL
总体积5ml
胶配好后混匀,灌胶2ml,插梳子时根据点样量和浓缩胶长短调整
梳子插入的深度,于室温(37℃)凝胶30min。
3.胶凝好后,拔梳子,将胶放入电泳槽中,加入1×Tris-Gly电泳缓冲液没过点样孔,胶外侧缓冲液界面应完全没过电极
4.Marker点5uL,用10uL小枪点样,以防样品冲出点样孔与旁边样品混杂。
电压打到70V,保证电流在20mA-30mA之间,当样品跑入浓缩胶时可适当调高电压,但不宜多次调整电压,否则条带会跑歪。
5.当溴酚蓝电泳至胶槽底部时,一般需要2.5h,停止电泳,剥胶,将浓缩胶部分切除后,加入考马斯亮蓝染色液,40r/min染色3h,可以过夜染色。
6.用移液器回收考马斯亮蓝染色液,再用蒸馏水将浮色冲洗干净后,倒入考马斯亮蓝脱色液,40r/min脱色至胶背景透明,期间可每2h更换一次脱色夜。
根据菌落SDS-PAGE的结果,可以看到目的蛋白是否大量表达。
2. 鉴定目的蛋白形成的是包涵体还是可溶性蛋白
1. 从冻存的菌液中取10ul,接种于5ml的LB培养基中,加入5ul
Amp(100 mg/ml),过夜活化菌株。
2. 按照1:50的比例,从活化的菌株中,取1ml菌液接种于50ml
LB培养基中,加入50ul Amp(100 mg/ml),扩大培养3 h。
3. 扩大培养后的菌液,取出10ml,不加IPTG,作为空白对照。
剩余的40ml菌液中加入40ul IPTG(0.5mol/l),诱导蛋白表
达。两者在37℃,200rpm的条件下培养3h。
4. 用50ml 离心管收集菌体,8000rpm,离心3min。注意配平。
5. 倒掉上清,加入40ml左右的dH2O洗菌体一遍,12000 rpm,
离心2min。注意要将菌体充分打散。
6. 倒掉上清,用枪头将上清吸干净。根据菌体的浓度,加入适
量的PBS buffe r 悬浮菌体。
7. 用大功率的超声波将菌体破碎,其间超声波每工作2min,停
止1min。菌体要始终保持在冰水混合物上,以保证低温,蛋
白不易变性。如此反复6-10遍,直至菌液清澈。
8. 12000rpm, 离心3min,分离上清和沉淀。
9. 在沉淀中加入250ul 1×SDS Loading buffer,取200u l 上清,
加入50ul 5×SDS Loading buffer,在恒温加热器上100℃加热
10min。
10.加热后的样品冷却到室温后,12000rpm,离心2min。
11.取上清40u l 点样,按照一中的方法进行SDS-PAGE检测。点样顺
序是:对照的上清,沉淀,样品的上清,沉淀。
12.根据SDS-PAGE结果,判断目的蛋白是以包涵体还是以可溶性蛋白
的形式产生。如果蛋白在沉淀中大量表达,则形成包涵体,需要
改变诱导表达条件,比如将扩大培养与诱导培养的温度改为
25℃,并延长相应培养时间。如果蛋白在上清中表达,则形成可
溶性蛋白,可继续进行下一步实验。
3. 纯化目的蛋白(整个过程要尽量保持在4℃进行)
1. 大量诱导目的蛋白(一般需要2-4 L的菌量),用超声波破碎
细胞,收集上清。
2. GST纯化柱的柱床保存在20%的乙醇中,使用之前,先用吸
管将柱床轻轻加入到柱子中,打开柱子,流出乙醇,使柱床
沉积下来。
3. 加入10ml PBS buffer,洗柱床3遍,使得柱床重新平衡。
4. 将准备好的样品加入到柱子中,每次加入5-10ml样品,使样
品结合在柱子上(每次上样量不能太多,因为柱子的结合能
力有限)。
5. 用PBS buffer洗柱子3遍,每次用10ml,去除柱子中非特异性
结合的杂蛋白。
6.
7.
8. 用分光光度计测每一管的OD值,记录下读数,将读数大于
200的样品留下(一般是第2至第4管),冻于-20℃冰箱中。
9. 重复步骤4-8,直至所有样品均纯化完毕。
10.可以用SDS-PAGE检测纯化出来的样品浓度以及纯度。
重生柱子的方法:(柱子使用三四次之后,需要重生,即让柱子上的GST结合基团重新暴露出来)
1.用还原型谷胱甘肽洗脱液洗柱子3遍,每次加入10ml。
2. 用 PBS缓冲液洗柱子4遍,每次加入10ml。
3. 用50 mmol Tris HCl(pH 8.0)洗柱子3遍,每次加入10ml。
4.用0.5M NaCl(pH 8.0)洗柱子3遍,每次加入10ml。
5. 用100 mM醋酸钠(pH 4.5)洗柱子3遍,每次加入10ml。
6. 用0.5M NaCL洗柱子3遍,每次加入10ml。
柱子长时间不用,需要按照以下方式清洗及保存:
1. 按照重生的1-6步骤先将柱子重生一遍。
2. PBS洗柱子2遍,每次加入10ml。
3. 将柱床用吸管吸出,分别用5%,10%,15%,20%的乙醇洗2
遍。
4. 用单蒸水轻轻冲洗柱子底部的膜,洗出杂蛋白。
5. 柱床保存在含20%乙醇的PA瓶中。柱子和柱床置于4度冰箱
保存。
如果柱子用于纯化不同的蛋白, 只需要按照重生方法的1,2步骤操作即可,然后直接纯化另外一个蛋白。如果柱子流速很慢,可能是柱子底部的膜被杂蛋白堵住了。柱子重生之后,将柱床用吸管吸出,然后用单蒸水将柱子底部的膜轻轻冲洗,再将柱床加入到柱子中,重新上样。
如果柱子需要重生,请用完柱子的同学自觉完成这份工作,以免影响下一位同学的使用。
4. 根据不同的用途浓缩蛋白
1. 若蛋白用于制作抗体
如果洗脱液用的是tris-bufferr配制的,需要用1/4的PBS buffer进行透析,然后在冷冻干燥机中冻干浓缩,因为公司最
终需要将目的蛋白溶于PBS buffer中。
如果洗脱液是用PBS buffer配制的,则直接在冷冻干燥机中冻干浓缩。
GST 纯化柱手册上建议的是用tris-bufferr配制洗脱液,但用PBS buffer配制的洗脱液可以将目的蛋白洗脱下来。可自己
选用。
2. 若蛋白用于活性检测
由于蛋白在tris-bufferr和PBS buffer中,均能保持活性,所以不论用哪种,都不需要透析,直接浓缩干燥即可。
浅谈原核表达的技巧 摘要:原核表达是表达外源基因常用的方法,具有操作简单、快捷,需时较短,表达产量高,适合工业化等优点。本文作者根据自己的实践经验,总结了原核表达的一些技巧。 关键词:原核表达表达载体限制性内切酶 将植物、动物、微生物等的目的基因插入合适载体后导入大肠杆菌用于表达大量蛋白质的方法一般称为原核表达。这种方法在蛋白纯化、定位及功能分析等方面都有应用。大肠杆菌用于表达重组蛋白有以下优点:易于生长和控制;易于培养,实验耗费少;可选择多种大肠杆菌菌株及与之匹配的具各种特性的质粒。原核表达是近年来表达外源蛋白常用的方法,本文根据自己的实践经验,着重谈谈对原核表达中的技巧问题。 一、原核表达一般程序 表达前准备-获得目的基因-构建含目的片段的表达载体(测序验证)-转化表达宿主菌-诱导靶蛋白的表达-表达蛋白的分析。 二、原核表达中各操作步骤的关键因素及技巧 1.表达前的准备要素:原核表达注重表达前对目的片段、表达载体及表达菌株的分析、选择。正所谓“磨刀不误砍柴功”,经过细致、周全的分析、准备、设计可带来较为顺当的实验,可免去许多不必要的麻烦。 (1)对表达载体的分析 载体的选择:同样的载体,同样的系统,很可能表达这个蛋白表达量起高,但另外一个就是做不出来,所以表达载体的选择非常重要,没有万能的载体。选择载体通常我们关心质粒上的几个功能组件及所带来的问题:是否为诱导表达型载体,启动子的强弱、多克隆位点、限制性内切酶的位置、终止密码子的有无及位置,融合Tag的有无,筛选报告基因的位置等。所选载体一定要保持原来的遗传背景(有些载体经过多次交换已变异)。选择表达载体时,要根据所表达蛋白的最终应用考虑,如果为了方便纯化,可选择融合表达;如果为了获得天然蛋白,可选择非融合表达。融合表达时在选择外源DNA同载体分子连接反应时,对转录和转译过程中密码结构的阅读不能发生干扰。 翻译的起始位点:要表达目的蛋白,在该基因的5’端必须有一起始位点,现在大部分的表达载体都提供起始位点,起始密码子与核糖体结合位点的距离都已被优化,一般情况下不需要自己再加,实际操作时要留意载体图谱上是否注明有起始密码子和终止密码子,如无,还得根据自己的实际情况加上。 在起始密码子附近的mRNA二级结构:外源基因其始转录后,保持mRNA的有效延伸、终止及稳定存在是外源基因有效表达的关键,尤其是在起始密码子附近的mRNA二级结构可能会抑制翻译的起始或者造成翻译暂停从而产生不完全的蛋白。如果利用Primer Premier软件分析DNA或RNA结构上有柄(stem)结构,并且结合长度超过8个碱基,这种结构会因为位点专一突变等因素而变得不稳定,影响正常的翻译。 (2)对目的片段的分析 基因(或蛋白)的大小:原核表达的成功与否与所要表达的蛋白(或基因)大小有关,一般说来小于5kD或者大于100kD的蛋白都是难以表达的。蛋白越小,越容易被内源蛋白水解酶所降解。在这种情况下可以采取串联表达,在每个表达单位(即单体蛋白)间设计蛋白水解或者是化学断裂位点。如果蛋白较小,那么加入融合标签GST、Trx、MBP或者其它较大的促进融合的蛋白标签就较有可能使蛋白正确折叠,并以融合形式表达。如果蛋白较大,大于60kD的蛋白建议使用较小的标签(如6×组氨酸标签)。对于结构研究较清楚的蛋白可以采取截取表达。当然表达时要根据目的进行截取,如果是要进行抗体制备而截取,那么一定要保证截取的部位抗原性较强。对于抗原性也可以利用软件分析,比如Vector NIT Suite或者一些在线软件,不过在分析之余也要认识到这是一种资料统计的结论,
原核表达遇到瓶颈怎么 办 TPMK standardization office【 TPMK5AB- TPMK08- TPMK2C- TPMK18】
我想表达遇到的第一个瓶颈估计就是为什么我的外源片段插到载体里面,PCR鉴定没问题,双酶切也OK,可是就是不见表达。一般我们是如何判断没有表达呢?大多都是首先进行SDS-PAGE。在跑胶的时候一定要设对照,比较严谨的电泳对照,应该是:Marker,标准品阳性对照(如果有,且打算做Western的话),以及空白载体(诱导)和重组载体(不诱导)2个阴性对照,再加上诱导不同时间的表达结果。Marker用于判断条带大小,标准品用于判断Western体系包括抗体显色剂和操作的可靠性,以及精确判断大小;空白载体(诱导)负对照有助于判断在非诱导条件下的本底表达;而重组载体(不诱导)负对照则有助于判断诱导的效果以及排除诱导剂对宿主菌潜在的干扰,或者是区分细菌内源和外源表达产物——偷懒可不是好习惯,会影响结果判断。注意,接种表达和接种做感受态都类似,一定要用挑单克隆、加足量抗生素、过夜培养的新鲜菌液转种最好,在筛选压力下生长旺盛的种子液远比经过4℃保存的菌液要好得多,也许是因为保存时间长会导致抗生素失效部分细菌丢失质粒或者其他变化。反正就是一种经验做法。 跑SDS-PAGE的话可以用考马斯亮兰染,它灵敏度在100ng左右;但是不能跟着做Western了。银染的灵敏度在0.1~1 ng;有钱还可以用Sypro Red,灵敏度高还可以继续做Western。WesternBlot是蛋白质定性和半定量的最通用的技术,具体细节可参考生物通WesternBlot技术专题,我们有详细介绍的。 在做过Western Blot仍没有检测到表达带,那么就要开始进行下一步的分析了。首先,看看载体的多克隆位点和片断插入的序列,是否有因为酶切连接而意外引入了转录终止信号。有时载体几经多个实验人员的周转,反复插入片断,或者是粘端
原核蛋白表达常见问题解析 1、为什么目的蛋白总是以包涵体的形式出现? 在原核蛋白表达纯化中目的蛋白经常发生错误的折叠,并聚集成为 包涵体。经过诱导,目的蛋白通常可达细胞总蛋白的50%以上。虽然有一定比例的蛋白以可溶的单体形式存在,而多达95%(甚至更多)的蛋白则在包涵体中。实验过程中,可以采取降低诱导温度,例如25–30°C,或降低IPTG浓度(0.01–0.1mM)并延长诱导时间,还有采用特别的 培养基等方法获得更多的可溶蛋白。 2、跨膜蛋白为什么很难表达? 跨膜蛋白的表达成功率相对较低是一个实验结果,究其原理,目前 众说纷纭很多种理论。以我们浅薄的理解层面来看,主要有以下几个 原因: 跨膜蛋白一般都是强疏水性的氨基酸分子和亲水性的分子跳跃式 的连接,形成的亲水疏水的一个最简单的跨膜化学结构,这种结构与 信号肽结构相似,对于原核细胞来说,简单的细胞器很难像真核细胞 一样完成信号肽识别及切除、引导内质网、高尔基体重新包装及分泌 这一复杂过程,有些蛋白是多次跨膜,对于原核细胞来说几乎是不可 能完成的任务。 另外,对于疏水性的片段,在原核细胞中极易形成包涵体,疏水 性多肽会抑制翻译过程,甚至与原核膜结构融合形成毒性,出于生物 自我保护的本能,所有的细胞器都会停止合成蛋白的过程。 3、如何选择蛋白表达宿主菌?
4、我们有哪些原核蛋白纯化方式?如何选择不同的纯化方式? 答:我们公司的蛋白纯化方法大致分为亲和纯化、离子交换、切胶 回收三类。 1、常规情况下,一般携带融合标签(His标签,GST标签,sumo标签,Fc标签),我们可以通过Ni柱、GST柱、Protein A等进行亲和纯化 获得融合蛋白,用亲和纯化的方法一般可以获得85%以上纯度的蛋白, 亲和纯化的方便快捷。 2、如果需要目的蛋白不含有任何标签,怎么选择纯化方式?。 (1)可表达融合蛋白,用蛋白工具酶切割融合蛋白,再进行纯化除去 工具酶。此方法能快速得到蛋白。 (2)可表达不含标签的蛋白,进行离子、分子筛、疏水等纯化,通过AKATA纯化设备获得蛋白。 3、如果需要获得蛋白作为抗原,可以直接通过切胶回收的方式,此方 法获得蛋白纯度较高,进行免疫动物后得到的抗体进行WB反应,灵敏 度较高。 5、表达得到的蛋白是有活性的么? 答:需要让蛋白有活性的条件很复杂,合适的缓冲液体系、盐浓度、蛋白的折叠状态甚至检测活性的方法的细微差别都可能导致活性 的强弱有无,一般情况下,上清表达的蛋白要比包涵体经过变复性纯 化后得到的蛋白活性要好,我们尽量从上清中获得蛋白,期许蛋白形 成的折叠最接近活性状态,这也是我们擅长的。但是在实际实验条件下,我们无法承诺表达纯化的蛋白一定具有客户期望的生理活性。
1.将已经成功转有重组表达载体pET-28a-CYP83A1的表达菌 E. coli.BL21(DE3)在LB固体培养基(50μg/mL Kan)上划线接种培 2.挑取单菌落,接种于5mL的LB液体培养基(50μg/mL Kan)中,37℃,180r/min振荡培养过夜。 3.取500μL过夜培养的菌液转接入100mL新的LB液体培养基(50μg/mL Kan)中,37℃,190r/min振荡培养到菌液OD600 =0.6~0.8。 4.分组培养:实验组加入终浓度为1mmol/L的IPTG,对照组不加IPTG,37℃,190r/min诱导培养6h。 5.8000r/min离心2min,收集细菌,用1×PBS(0.01mol/L)缓冲液悬浮。 6.冰上超声波破碎,功率30w,工作5s,间歇5s,总时间2min。 7.4℃、12000r/min离心10min,分离上清与沉淀,取100μL上清与等体积的2×上样缓冲液混合;用200μL1×上样缓冲液悬浮沉淀,沸水浴5min后,对上清和沉淀进行SDS-PAGE检测。
重组质粒在大肠杆菌中的诱导表达及SDS-PAGE分析 挑测序正确的单克隆接种到3mLLB(50μg·mL-1Kan)培养基中,振荡培养12h后,将菌液按1﹕100的比例加入到300mL LB(50μg·mL-1Kan)培养基中200r/min37℃振荡培养至OD600为0.5—0.6时,加入IPTG(使终浓度为1mmol·L-1)进行诱导表达,分别在37℃诱导4h,4℃保存备用。未加IPTG诱导的pET-28a-CsFOMT收集作为阴性对照。诱导全部完成后,各取50mL 菌液离心收集细菌,加入SDS上样缓冲液,悬浮混匀,100℃3min,12000r/min离心1min,取上清4℃保存备用。另取50mL菌液离心收集菌体后用1×PBS(PH7.4)将沉菌悬起,经过超声波细胞破碎(20mm的变幅杆,400W,超声2s,间隔5s,重复60次),10000r/min离心10min分离上清和沉淀,上清和沉淀样品中分别加入SDS上样缓冲液,混匀,沸水浴,取上清和沉淀分别进行SDS-PAGE(5%浓缩胶,12%分离胶),然后分析蛋白表达结果
载体与表达系统 0001--pIRES --BD Co--真核表达--yshu3507 0002--pECFP-C1--BD Co--真核表达--yshu3507 0003--pShuttle--/--/--victoh 0004--pSBR322--/--/--chujun_hust 0005--pcDNA3.1(+)/CAT--invitrogen--真核表达--wangjun2002274 0006--pQEx--Qiagen--原核表达--wangjun2002274 0007--pIVEX2.3--Roche--体外转录翻译--wangjun2002274 0008--pIRES-EGFP--/--/--luoqingli2003 0009--pET-28a(+)--Novagen--原核表达--wangjun2002274 0010--pSUPER Vector--oligoengine--siRNA--zyh961171 0011--pET-32a(+)--novagen--原核表达--Crazyvirus 0012--pBK-CMV--/--原核表达、真核表达--zrzhong6519 0013--pcDNA3.1/Zeo (+)--invitrogen--原核表达--Crazyvirus 0014--pcDNA3--invitrogen--真核表达--kras 0015--pfastbac1--invitrogene--昆虫表达--cox2wj 0016--pEGFP-C3--clontech--真核表达--smilely 0017--pSecTag2--invitrogen--真核表达--kenmed 0018--PYX212--/--真核表达--Gmail 0019--pET20b--Novagen--原核表达--Gmail 0020--pEGFP-1--BD BIO--转录调控--wyh 0021--pEGFP/U6 --/--siRNA--songbinn
原核表达 一、原理 1、E . coli 表达系统 E . coli 是重要的原核表达体系。在重组基因转化入E . coli 菌株以后,通过温度的控制,诱导其在宿主菌内表达目的蛋白质,将表达样品进行SDS-PAGE 以检测表达蛋白质。 2、外源基因的诱导表达 提高外源基因表达水平的基本手段之一,就是将宿主菌的生长与外源基因的表达分成两个阶段,以减轻宿主菌的负荷。常用的有温度诱导和药物诱导。本实验采用异丙基硫代-β-D-半乳糖昔(IPTG)诱导外源基因表达。 不同的表达质粒表达方法并不完全相同,因启动子不同,诱导表达要根据具体情况而定。 二、材料 1、诱导表达材料 ( 1 ) LB (Luria—Bertani))培养基 酵母膏(Yeast extract) 5g 蛋白胨(Peptone) 10g NaCl 10g 琼脂(Agar) 1-2% 蒸馏水(Distilled water) 1000ml pH 7.0 适用范围:大肠杆菌 ( 2 ) IPTG 贮备液:2 g IPTG溶于10 mL 蒸馏水中,0 . 22 μm 滤膜过滤除菌,分装成1 mL /份,-20 ℃保存。 ( 3 ) l×凝胶电泳加样缓冲液: 50 mmol / L Tris -CI ( pH 6 . 8 ) 50 mmol / L DTT 2 % SDS (电泳级) 0.1 %溴酚蓝 10 %甘油 2、大肠杆菌包涵体的分离与蛋白纯化材料 1 )酶溶法 (1)裂解缓冲液: 50 mmol / L Tris-CI ( pH 8 . 0 ) 1 mmol / L EDTA 100 mmol / LNaCI (2)50 mmol / L 苯甲基磺酰氟(PMSF )。 (3)10 mg / mL 溶菌酶。 (4)脱氧胆酸。 (5)1 mg / mL DNase I。 2 )超声破碎法 ( 1 ) TE 缓冲液。 ( 2 ) 2×SDS -PAGE 凝胶电泳加样缓冲液: 100 mmol / L Tris-HCI ( pH 8 . 0 ) 100 mmol / L DTT 4 %SDS 0.2 %溴酚蓝 20 %甘油
原核、真核表达载体构建 真核表达载体和原核表达载体的区别:主要是因为原核和真核表达系统所需的表达元件不同。 比如说启动子,终止子在两种表达系统中是不一样的。 带有真核表达元件的是真核载体,能在真核生物内表达; 带有原核表达元件的是原核载体,能在原核生物内表达。两者都具有的为穿梭载体。 ㈠原核表达载体指:能携带插入的外源核酸序列进入原核细胞中进行复制的载体。 原核表达载体调控原件 1.启动子 启动子是DNA链上一段能与RNA聚合酶结合并起始RNA合成的序列,它是基因表达不可缺少的重要调控序列。没有启动子,基因就不能转录。由于细菌RNA聚合酶不能识别真核基因的启动子,因此原核表达载体所用的启动子必须是原核启动子。原核启动子是由两段彼此分开且又高度保守的核苷酸序列组成,对mRNA的合成极为重要。在转录起始点上游5~10 bp处,有一段由6~8个碱基组成,富含A和T的区域,称为Pribnow 盒,又名TATA 盒或-10区。来源不同的启动子,Pribnow 盒的碱基顺序稍有变化。在距转录起始位点上游35 bp 处,有一段由10 bp组成的区域,称为-35区。转录时大肠杆菌RNA聚合酶识别并结合启动子。-35区与RNA聚合酶s亚基结合,-10区与RNA聚合酶的核心酶结合,在转录起始位点附近DNA被解旋形成单链,RNA聚合酶使第一和第二核苷酸形成磷酸二酯键,以后在RNA聚合酶作用下向前推进,形成新生的RNA 链。原核表达系统中通常使用的可调控的启动子有Lac(乳糖启动子)、Trp(色氨酸启动子)、Tac(乳糖和色氨酸的杂合启动子) 、lPL (l噬菌体的左向启动子)、T7噬菌体启动子等。 2. SD序列 1974年Shine和Dalgarno首先发现,在mRNA上有核糖体的结合位点,它们是起始密码子AUG和一段位于AUG上游3~10 bp处的由3~9 bp组成的序列。这段序列富含嘌呤核苷酸,刚好与16S rRNA 3¢末端的富含嘧啶的序列互补,是核糖体RNA的识别与结合位点。以后将此序列命名为Shine-Dalgarno序列,简称SD序列。它与起始密码子AUG之间的距离是影响mRNA转录、翻译成蛋白的重要因素之一,某些蛋白质与SD序列结合也会影响mRNA与核糖体的结合,从而影响蛋白质的翻译。另外,真核基因的第二个密码子必须紧接在ATG 之后,才能产生一个完整的蛋白质。 3.终止子 在一个基因的3¢末端或是一个操纵子的3'末端往往有特定的核苷酸序列,且具有终止转录功能,这一序列称之为转录终止子,简称终止子(terminator)。转录终止过程包括:RNA聚合酶停在DNA模板上不再前进,RNA的延伸也停止在终止信号上,完成转录的RNA从RNA聚合酶上释放出来。对RNA聚合酶起
如何做原核表达 人们合成与生物相关的物质是从尿素开始的,1828年,德国化学家维勒人工合成了存在于生物体的这种有机物。在1960年我国科学家采用化学方法首次成功地合成了具有生物活性的蛋白质——胰岛素。随着内切酶的发现和基因工程技术的发展,人们发现用各种不同的载体在原核、真核系统中进行蛋白表达更为行之有效。而这其中大肠杆菌表达系统发展得最为迅速、成熟。原核表达具有操作方便、快捷,需时较短,表达量大,适合工业化生产等优点。虽然也有缺少糖基化和表达后加工等问题,当有了其它多种表达系统后,原核系统仍是我们合成外源蛋白的首选。 在网上看到有人把原核表达技术分成四个等级:初次尝试扫盲、乱棍打枣入门、系统优化中级和自成一体高手,觉得十分有意思。但是根据笔者自己的经验以及耳闻目睹的一些经历告诉我:做表达?那是谋事在人,成事在天。有时候你把克隆做出来了,双酶切鉴定没问题,测序没问题,可是就是看不到表达带。原因当然可以分析,实验也是可以改进,但是窜改一下戈尔泰的话:“成功的实验都是一样的,失败的实验各有各的不幸。”在实验遇到瓶颈的时候要如何进行分析,找到问题的症结是我们的实验关键所在。在准备进行原核表达的时候需要考虑的因素很多,市面上可供选择的载体、菌株也很多,要如何进行正确的选择,找到适合自己的载体是十分重要的。所以,现在要对目前常用的一些载体进行介绍,让我们对其相关产品及其表达原理进行了解,以方便实验设计。 首先来一些大肠杆菌表达的基本概念:一个完整的表达系统通常包括配套的表达载体和表达菌株,如果是特殊的诱导表达还包括诱导剂,如果是融合表达还包括纯化系统或者Tag检测等等。选择表达系统通常要根据实验目的来考虑,比如表达量高低,目标蛋白的活性,表达产物的
真核细胞表达系统的类型与常用真核细胞表达载体 标签:真核细胞酵母表达系统细胞表达载体真核表达系统昆虫表达系统动物表达系统 摘要: 原核表达系统是常被用来研究基因功能的成熟系统,由于原核表达系统具有包涵体蛋白不易纯化、蛋白修饰不完整等缺陷,人们也开始利用真核细胞表达系统来研究基因。 原核表达系统是常被用来研究基因功能的成熟系统,由于原核表达系统具有包涵体蛋白不易纯化、蛋白修饰不完整等缺陷,人们也开始利用真核细胞表达系统来研究基因。 自上世纪70年代基因工程技术诞生以来,基因表达技术已渗透到生命科学研究的各个领域。并随着人类基因组计划实施的进行,在技术方法上得到了很大发展,时至今日已取得令人瞩目的成就。随着人类基因组计划的完成,越来越多的基因被发现,其中多数基因功能不明。利用表达系统在哺乳动物细胞内表达目的基因是研究基因功能及其相互作用的重要手段。 在各种表达系统中,最早被采用进行研究的是原核表达系统,这也是目前掌握最为成熟的表达系统。该项技术的主要方法是将已克隆入目的基因DNA段的载体(一般为质粒)转化细菌(通常选用的是大肠杆菌),通过iptg诱导并最终纯化获得所需的目的蛋白。其优点在于能够在较短时间内获得基因表达产物,而且所需的成本相对比较低廉。但与此同时原核表达系统还存在许多难以克服的缺点:如通常使用的表达系统无法对表达时间及表达水平进行调控,有些基因的持续表达可能会对宿主细胞产生毒害作用,过量表达可能导致非生理反应,目的蛋白常以包涵体形式表达,导致产物纯化困难;而且原核表达系统翻译后加工修饰体系不完善,表达产物的生物活性较低。 为克服上述不足,许多学者将原核基因调控系统引入真核基因调控领域,其优点是: ①根据原核生物蛋白与靶DNA间作用的高度特异性设计,而靶DNA与真核基因调控序列基本无同源性,故不存在基因的非特异性激活或抑制; ②能诱导基因高效表达,可达105倍,为其他系统所不及; ③能严格调控基因表达,即不仅可控制基因表达的“开关”,还可人为地调控基因表达量。 因此,利用真核表达系统来表达目的蛋白越来越受到重视。目前,基因工程研究中常用的真核表达系统有酵母表达系统、昆虫细胞表达系统和哺乳动物细胞表达系统。 1.酵母表达系统 最早应用于基因工程的酵母是酿酒酵母,后来人们又相继开发了裂殖酵母、克鲁维酸酵母、甲醇酵母等,其中,甲醇酵母表达系统是目前应用最广泛的酵母表达系统。目前甲醇酵母主要有H Polymorpha,Candida Bodini,Pichia Pastris3种。以Pichia Pastoris应用最多。
Chi l 原核表达基本试验步骤 将克隆化基因插入合适载体后导入大肠杆菌用于表达大量蛋白质的方法一般称为原核表达。这种方法在蛋白纯化、定位及功能分析等方面都有应用。大肠杆菌用于表达重组蛋白有以下特点:易于生长和控制;用于细菌培养的材料不及哺乳动物细胞系统的材料昂贵;有各种各样的大肠杆菌菌株及与之匹配的具各种特性的质粒可供选择。但是,在大肠杆菌中表达的蛋白由于缺少修饰和糖基化、磷酸化等翻译后加工,常形成包涵体而影响表达蛋白的生物学活性及构象。 表达载体在基因工程中具有十分重要的作用,原核表达载体通常为质粒,典型的表达载体应具有以下几种元件: (1)选择标志的编码序列; (2)可控转录的启动子; (3)转录调控序列(转录终止子,核糖体结合位点); (4)一个多限制酶切位点接头; (5)宿主体内自主复制的序列。 原核表达一般程序如下:获得目的基因-准备表达载体-将目的基因插入表达载体中(测序验证)-转化表达宿主菌-诱导靶蛋白的表达-表达蛋白的分析-扩增、纯化、进一步检测,其中包括: 一、试剂准备 (1)LB培养基。 (2)1M IPTG(异丙基硫代-β-D-半乳糖苷):2.38g IPTG溶于10ml ddH2O
中,0.22μm滤膜抽滤,-20℃保存。 CCY的IPTG是1M的,用时进行1000倍稀释。 二、操作步骤 (一)获得目的基因 1、通过PCR方法:以含目的基因的克隆质粒为模板,按基因序列设计一对引物(在上游和下游引物分别引入不同的酶切位点),PCR循环获得所需基因片段。 2、通过RT-PCR方法:用TRIzol法从细胞或组织中提取总RNA,以mRNA 为模板,逆转录形成cDNA第一链,以逆转录产物为模板进行PCR循环获得产物。 (二)构建重组表达载体 1、载体酶切:将表达质粒用限制性内切酶(同引物的酶切位点)进行双酶切,酶切产物行琼脂糖电泳后,用胶回收Kit或冻融法回收载体大片段。 2、PCR产物双酶切后回收,在T4DNA连接酶作用下连接入载体。我们用Soultion I连接。 (三)获得含重组表达质粒的表达菌种 1、将连接产物转化大肠杆菌BL21,根据重组载体的标志(抗Amp或蓝白斑)作筛选,挑取单斑,碱裂解法小量抽提质粒,双酶切初步鉴定。 2、测序验证目的基因的插入方向及阅读框架均正确,进入下步操作。否则应筛选更多克隆,重复亚克隆或亚克隆至不同酶切位点。 3、以此重组质粒DNA转化表达宿主菌的感受态细胞。
原核表达步骤
Chi l 原核表达基本试验步骤 将克隆化基因插入合适载体后导入大肠杆菌用于表达大量蛋白质的方法一般称为原核表达。这种方法在蛋白纯化、定位及功能分析等方面都有应用。大肠杆菌用于表达重组蛋白有以下特点:易于生长和控制;用于细菌培养的材料不及哺乳动物细胞系统的材料昂贵;有各种各样的大肠杆菌菌株及与之匹配的具各种特性的质粒可供选择。但是,在大肠杆菌中表达的蛋白由于缺少修饰和糖基化、磷酸化等翻译后加工,常形成包涵体而影响表达蛋白的生物学活性及构象。 表达载体在基因工程中具有十分重要的作用,原核表达载体通常为质粒,典型的表达载体应具有以下几种元件: (1)选择标志的编码序列; (2)可控转录的启动子; (3)转录调控序列(转录终止子,核糖体结合位点); (4)一个多限制酶切位点接头; (5)宿主体内自主复制的序列。 原核表达一般程序如下:获得目的基因-准备表达载体-将目的基因插入表达载体中(测序验证)-转化表达宿主菌-诱导靶蛋白的表达-表达蛋白的分析-扩增、纯化、进一步检测,其中包括: 一、试剂准备 (1)LB培养基。 (2)1M IPTG(异丙基硫代-β-D-半乳糖苷):2.38g IPTG溶于10ml ddH2O
中,0.22μm滤膜抽滤,-20℃保存。 CCY的IPTG是1M的,用时进行1000倍稀释。 二、操作步骤 (一)获得目的基因 1、通过PCR方法:以含目的基因的克隆质粒为模板,按基因序列设计一对引物(在上游和下游引物分别引入不同的酶切位点),PCR循环获得所需基因片段。 2、通过RT-PCR方法:用TRIzol法从细胞或组织中提取总RNA,以mRNA 为模板,逆转录形成cDNA第一链,以逆转录产物为模板进行PCR循环获得产物。 (二)构建重组表达载体 1、载体酶切:将表达质粒用限制性内切酶(同引物的酶切位点)进行双酶切,酶切产物行琼脂糖电泳后,用胶回收Kit或冻融法回收载体大片段。 2、PCR产物双酶切后回收,在T4DNA连接酶作用下连接入载体。我们用Soultion I连接。 (三)获得含重组表达质粒的表达菌种 1、将连接产物转化大肠杆菌BL21,根据重组载体的标志(抗Amp或蓝白斑)作筛选,挑取单斑,碱裂解法小量抽提质粒,双酶切初步鉴定。 2、测序验证目的基因的插入方向及阅读框架均正确,进入下步操作。否则应筛选更多克隆,重复亚克隆或亚克隆至不同酶切位点。 3、以此重组质粒DNA转化表达宿主菌的感受态细胞。
原核生物和真核生物基因表达调控特点的比较1.相同点:转录起始是基因表达调控的关键环节2.不同点:A.原核基因的表达调控主要包括转录和翻译水平 真核基因的表达调 控主要包括染色质活化、转录、转录后加工、翻译、翻译后加工多个层次B.原核基因表达调控主要为负调控,真核主要为正调控C.原核转录不需要转录因子,RNA聚合酶直接结合启 动子,由sita因子决定基因表的的特异性 真核基因转录起始需要基础特异两类转录因子 依赖DNA-蛋白质、蛋白质-蛋白质相互作用 调控转录激活D.原核基因表达调控主要采用操纵子模型 转录出多顺反子RNA 实现协调调节 真核基因转录产物为单顺反子RNA 功能相关蛋白的协调表达机制更为复杂。真核生物基因表达调控的环节主要在转录水平 其次是翻译水平。原核生物基因以操纵子的形式存在。转录水平调控涉及到启动子、sita因子 与RNA聚合酶结合 、阻遏蛋白 负调控 、正调控蛋白、倒位蛋白、RNA聚合酶抑制物、衰减子等。翻译水平的调控涉及SD序列、mRNA的稳定性 不稳定(5’端和3’端的发夹结构可保护不被酶水解mRNA的5’端与核糖体结合 可明显提高稳定性)、翻译产物及小分子RNA的调控作用。真核生物基因表达的调控环节较多 在DNA水平上可以通过染色体 丢失、基因扩增、基因重排、DNA甲基化、染色体结构改变影响基因表达。在转录水平主要通过反式作用因子调控转录因子与TATA盒的结合、RNA聚合酶与转录因子-DNA复合物的结合及转录起始复合物的形成。在转录后水平主要通过RNA修饰、剪接及mRNA运输的控制来影响基因表达。在翻译水平有影响起始翻译的阻遏蛋白、5’AUG、5’端非编码区长度、mRNA 的稳定性调节及小分子RNA。真核基因调控中最重要的环节是基因转录 真核生物基因表达需要转录因子、启动子、沉默子和增强子。葡萄糖存在 乳糖不存在 此时无诱导剂
原核表达详细步骤 PartⅠ选择表达的目的基因 一、基因序列 1. 得到靶基因DNA(cDNA)序列,有几种方式寻找正确的读码顺序: ①利用生物信息学在NCBI上blast同源基因,找到同源蛋白,再在DNA 的ORF中找到正确的读码。 ②实验方法,即得到蛋白,进行测序,然后在DNA上找到正确的读码。 ③利用mRNA的特征,找到启动子,编码区,终止子。在编码区中找到翻译起始密码子与终止密码子(cDNA)。 2. 注意事项: ①区别ORF和CDS→ORF一般在DNA上的定义,寻找原则是翻译起始密码子和终止密码子;CDS可以是DNA上的定义,也可以是mRNA上的定义,分为complete CDS和partial CDS,是从第一个核酸开始读,连续读下去,complete CDS读码是“M、、、、、、、、、*”,partial CDS的读码是相应的AA ②在进行试验设计时,充分利用生物信息学的信息后,在进行试验设计。 二、抗原决定簇的预测 1、原理: 蛋白质表面部分可以使免疫系统产生抗体的区域叫抗原决定簇。一般抗原决定簇是由6-12 氨基酸或碳水基团组成,它可以是由连续序列(蛋白质一级结构)组成或由不连续的蛋白质三维结构组成。变性蛋白只是天然蛋白伸直的了产物,用来免疫动物具有更强的抗原性。只是天然蛋白中被包在内部的抗原决定簇也会暴露出来,如果用该变性抗原制备的抗体来检测变性抗原是可以的,如果用来检测天然蛋白,可能会有假阳性。做单抗也可以,同样道理,筛选出的单抗可能对抗的抗原决定簇处于天然抗原的内部,是否能用还要看将来该单抗用来干什么。 2、选择原则: (1)、亲水性:大部分抗原决定簇是亲水性的。
原核表达步骤 原核表达先要将基因克隆到原核表达载体上,然后通过转化到 JM109或BL21等菌株中,诱导表达蛋白,然后进行蛋白纯化。本实验方案的前提是,目的基因已克隆到载体,并已转进入JM109菌株中。 1.鉴定目的蛋白是否在大肠杆菌JM109或BL21中大量表达 (1)制样 1 . 挑取经过双酶切鉴定的单克隆菌落于700ul LB培养基,加入0.7ul Amp(100mg/mL),37o C200r/min摇床培养,过夜活化。 2. 以1:50比例(200ul),将活化的过夜培养物加入10mL LB液体培养基中,加入10uLAmp(100mg/ml),37o C200r/min摇床扩大培养2h-3h,期间取样监控菌液的OD值,控制菌液OD600在0.6-1.0之间,以使大肠杆菌处于最适合表达外源蛋白的生长状态。(一般3h时,菌液浓度及达到标准,但是不同的基因对菌的影响不同,所以第一次实验时需要确定这个最佳时间) 3. 从10ml扩大培养物中取3ml菌液作为不加IPTG的空白对照(CK),其余7ml菌液加入7ul IPTG(储存浓度为0.5mol/l),使IPTG 终浓度达到0.5mmol/l。以200r/min的转速,37o C摇床培养3h。 4. 以5000r/min离心2min收集菌体,倾倒上清,每个离心管收集3ml培养物。 5. 加入1ml dH2O,将管底沉淀用振荡器打散以充分洗涤,8000r/min 离心2min,倾倒上清。 6. 重复步骤5。将离心管中的水倒干净。 (二)菌落SDS-PAGE 1. 在收集的菌体中加入200ul 1×SDS PAGE loading buffer(可根据沉淀的量增加或减少loading buffer的量,一般200ul比较合适)。用漩涡器剧烈震荡,确保将管底沉淀震散。 2. 将样品于100℃恒温加热器上开盖加热10min(Marker也要加热)。样品凉后,12000r/min离心3min,取每管的上清点样。上样量一般30ul—40ul,marker 20ul。 (3)SDS-PAGE分析 1. 根据目的片段的大小,制作不同浓度的分离胶 蛋白分子量 (kDa)凝胶浓度 (%) 4-4020
原核表达和真核表达 原核表达的步骤和主要试剂 1、获得目的基因; 2、构建重组表达载体; 3、获得含重组表达质粒的表达菌种; 4、诱导表达; 5、包涵体的分离。 获得目的基因 PCR法钓基因 引物(捷瑞),普通taq酶(Regene,Fermentas)/Pfu 酶(Fermentas)/Hotstart Taq酶 (Fermentas),dNTP(Regene,Fermentas) RT-PCR法获取基因 Trizol(Invitrogen,捷瑞),DEPC(捷瑞或其他进分), Rnase抑制剂(MBI),M-MLV/AMV(Fermentas)或 M-MLV/AMV一步法RT-PCR试剂盒(捷瑞、Qiagen、 Axygen、Omega) 构建重组表达载体 内切酶(Fermentas),载体(捷瑞,Fermentas),琼脂糖(西班牙),T4连接酶(Fermentas,promega),测序 获得含重组表达质粒的表达菌种 菌株(捷瑞),质粒小抽试剂盒(捷瑞、Qiagen、Axygen),感受态细胞制备试剂盒(捷瑞),抗生素(捷瑞,Amresco,sigma),测序(伟沃) 诱导表达 IPTG(捷瑞,Amresco,sigma),胰蛋白胨(Oxiod),酵母粉(Oxiod),琼脂(进分,国产) 包涵体的分离 Triton X -100(捷瑞,进分),PMSF(sigma),DTT (Amresco,进分),尿素(Amresco,进分,sigma),还原型谷胱甘肽(Amresco,进分,sigma) 真核表达 蛋白质在真核表达的过程中,可以通过真核细胞的转录加工系统获得具有一定构象和生物活性的蛋白质。真核表达现在主要有毕赤酵母表达体系、杆状病毒昆虫细胞表达体系、哺乳动物细胞表达体系。 各种表达系统各有其优缺点,酵母和昆虫细胞表达系统蛋白表达水平高,生产成本低,但它们的加工修饰体系与哺乳动物细胞不完全相同;哺乳动物细胞产生的蛋白质更接近于天然蛋白质,但其表达量低、操作繁琐。各种表达系统由于翻译后的加工不完全相同,因而产生的重组蛋白的生物学活性和免疫原性有时会有差别。 毕赤酵母表达体系步骤及试剂 1、目的基因的获取; 2、构建表达质粒载体; 3、表达质粒的转化; 4、转化子的鉴定; 5、表达产物的鉴定。 目的基因的获取 基因组DNA提取 酚/氯仿法 蛋白酶K(Merk和Amresco),饱和酚(捷瑞), 氯仿﹕异戊醇(24﹕1)(捷瑞) 试剂盒法 血液/动物/植物/微生物基因组DNA提取试剂盒 (捷瑞、Qiagen、Axygen、Omega) RNA的提取 Trizol法 Trizol(Invitrogen,捷瑞),DEPC(捷瑞或其 他进分),Rnase抑制剂(MBI)
实验方法与步骤 1 表达质粒的构建及测序分析 1.1 cofilin-1的片段的准备 1.1.1 引物设计 根据在GenBank上查找人源cofilin-1的基因序列,用Primer Premier 5.0软件进行上下游引物的设计,并送往上海生物工程技术服务有限公司合成的PCR 引物。引物如下: 引物名称序列 F-cofilin-1 5′-AAGTCGACATATGGCCTCCGGTGTG-3′ R-cofilin-1 5′-TCTCTCGAGGGCTCACAAAGGCTTG-3′将以上引物用灭菌的三蒸水稀释成10μmol/L,分装于Eppendorf管中,-20℃冰箱中保存备用。 1.1.2 cofilin-1片段PCR 1 反应体系: 2.5μl KOD polymerase(3’-5’核酸外 切酶活性) KOD polymerase buffer 5μl MgSO4 2.5μl DMSO(“万能溶剂”) 2.5μl dNTPMixture 5μl PrimerF(底物) 1.5μl PrimerR 1.5μl Template(模板)5μl ddH2O 25μl Total 50μl 2PCR反应条件:
①94℃预变性3min ②94℃退火30s ③65℃延伸40s ④68℃40s ⑤go to②30个循环 ⑥68℃5min ⑦4℃forever 3 琼脂糖凝胶电泳对PCR产物进行检测 (1)配置浓度为1%的凝胶。称取琼脂糖0.3g,加入30ml 1×TAE电泳缓冲液(Tris-乙酸电泳缓冲液)中,用微波炉加热2min,待凝胶稍冷却,加入2μl EB(溴化乙锭,荧光染色剂)混匀后倾入凝胶铸槽中,插入梳子,并用玻璃棒驱除气泡,待凝胶完全凝结后拔除梳子。 (2)把凝胶置于1×TAE电泳缓冲液的电泳槽中,加样孔置于负极一侧,然后依次在加样孔中加入50μl Marker、50μl样品+10μl loading buffer(上样缓冲液,可以显示两条带,前面的蓝色的条带是溴酚蓝,代表的片段大小是300bp,后面的有点绿色的条带是二甲苯青,代表的片段大小在4000bp左右),盖上电泳盖,以100V电压进行电泳。 (3)当Marker条带充分分开后即可停止电泳,将凝胶移至保鲜膜上,置于凝胶自动成像仪中分析。 4 割胶回收PCR反应体系的扩增产物,用Omega Bio-Tek公司的Gel Extrection Kit进行回收: (1)将PCR扩增产物经1%的琼脂糖凝胶电泳,在紫外灯下迅速切取含有目的条带的琼脂糖凝胶,放入灭菌的EP管中。DNA在紫外灯下曝光时间不超过30s。 (2)称量凝胶块重量,以1g=1ml进行计算,加入适量体积的binding buffer,55-65℃加热至凝胶完全融化(约7~10min),每隔2~3min震荡一次。 (3)将Hibind DNA柱子套在2ml的收集管。将上述步骤(2)的溶液转移至Hibind DNA柱子中,10000×g离心1min,弃滤液。 (4)将柱子装回收集管中,加入300μl binding buffer,10000×g离心1min,
蛋白质的表达、分离、纯化实验 标签: 蛋白质 表达 分离 纯化 蛋白质表达、分离、纯化可以:(1)探索和研究基因的功能以及基因表达调控的机理;(2)供作结构与功能的研究;(3)作为催化剂、营养剂等。 详细 实验方法 原核表达法 实验方法原理 携带有目标蛋白基因的质粒在大肠杆菌BL21中,在 37℃,IPTG 诱导下,超量表达携带有6个连续组氨酸残基的重组氯霉素酰基转移酶蛋白,该蛋白可用一种通过共价偶连的次氨基三乙酸(NTA )使镍离子(Ni 2+)固相化的层析介质加以提纯,实为金属熬合亲和层析(MCAC )。蛋白质的纯化程度可通过聚丙烯酰胺凝胶电泳进行分析。 实验材料 大肠杆菌BL21 试剂、试剂盒 LB 液体培养基氨苄青霉素Washing BufferElution BufferIPTG 蒸馏水胰蛋白胨酵母粉氯化钠 仪器、耗材 摇床离心机层析柱离心管移液枪枪头盒烧杯玻璃棒 实验步骤 一、试剂准备 1. LB 液体培养基:Trytone 10 g , yeast extract 5 g ,NaCl 10 g ,用蒸馏水配至1000 mL 。 2. 氨苄青霉素:100 mg/mL 。 3. 上样缓冲液:100 mM NaH 2PO 4,10 mM Tris ,8M Urea ,10 mM 2-ME , pH8.0。 4. Washing Buffer :100 mM NaH 2PO 4,10 mM Tris ,8 M Urea ,pH6.3。 5. Elution Buffer :100 mM NaH 2PO 4,10 mMTris ,8M Urea , 500 mM
真核基因和原核基因表达调控的异同? 真核基因表达调控的基本原理与原核基因相同,主要表现在: 1、与原核基因的调控一样,真核基因表达调控也以转录水平调控为最重要; 2、在结构基因均有调控序列,并依靠特异蛋白因子与这些调控序列的结合与否调控基因的表达。 3、都要经历转录、翻译的过程。 4、表达过程都有复杂性,多环节 不同 1、真核基因表达调控过程更复杂。 2、在染色质结构上。原核细胞的DNA是裸露的,而真核细胞DNA包装在染色体中。DNA与组蛋白组成核小体形成为染色体基本单位。在原核细胞中染色质结构对基因的表达没有明显的调控作用,而在真核细胞中染色质的变化调控基因表达,并且基因分布在不同的染色体上,存在染色体间基因的调控问题; 3、真核生物中编码蛋白质的基因通常是断裂基因,含有有非编码序列即内含子,因而转录产生的mRNA前体必须剪切加工才能成为有功能的成熟的mRNA,而不同拼接方式的可产生不同的mRNA。而原核生物的基因由于不含有外显子和内含子,因此,转录产生的信使RNA不需要剪切、拼接等加工过程。 4、在原核基因转录的调控中,既有正调控,也有负调控,二者同等重要,而真核细胞中虽然也有正调控成分和负调控成分,但目前已知的主要是正调控,且一个真核基因通常都有多个调控序列,必须有多个激活物同时特异地结合上去才能调节基因的转录; 5、原核基因的转录和翻译通常是相互偶联的,而真核基因的转录与翻译在时空上是分开的,从而使真核基因的表达有多种调控机制。 6、真核生物细胞中存在mRNA的稳定性调控
7、真核生物大都为多细胞生物,基因的表达随细胞内外环境条件的改变和时间程序在不同的表达水平上进行着精确调控,而原核生物主要受环境因素和营养状况影响基因调控。 8、真核生物由三种RNA聚合酶分别负责三种RNA的转录,而原核生物只有一种。
[Merck推荐]原核表达秘笈之宿主菌株选择指南 在原核蛋白表达过程中,选择构建一个合适原核表达体系需要综合考虑3大因素:表达载体、宿主菌株、表达诱导条件,以获得最满意的表达效果。 事实上,在平时的实验中,最容易被忽视的就是宿主菌的选择——多数人会直接选择自己实验室曾经用过的表达菌株,或者是载体配套的菌株,而不去追究原因——即使表达结果不佳,大多在表达条件和载体上找原因,也不会考究菌株的选择是否适合。 作为原核表达的宿主,对外源基因的表达会产生一定的影响,是勿庸置疑的。每一个宿主细胞都像一个微观的小工厂,按照细胞固有的程序完成“你给它们安排的生产任务”——因为很难亲眼观察微观世界中表达是如何进行的,当出现问题时,我们需要经验判断问题所在。宿主细胞对原核表达可能会产生哪些影响呢? 知其然还要知其所以然。比如,菌株内源的蛋白酶过多,可能会造成外源表达产物的不稳定,所以一些蛋白酶缺陷型菌株往往成为理想的起始表达菌株。堪称经典的BL21系列就是lon和ompT蛋白酶缺陷型,也是我们非常熟悉的表达菌株。大名鼎鼎的BL21(DE3)融源菌则是添加T7聚合酶基因,为T7表达系统而设计。 真核细胞偏爱的密码子和原核系统有不同,因此,在用原核系统表达真核基因的时候,真核基因中的一些密码子对于原核细胞来说可能是稀有密码子,从而导致表达效率和表达水平很低。改造基因是比较麻烦的做法,Rosetta 2系列就是更好的选择——这种携带pRARE2质粒的BL21衍生菌,补充大肠杆菌缺乏的七种(AUA, AGG, AGA, CUA, CCC, GGA 及CGG)稀有密码子对应的 tRNA,提高外源基因、尤其是真核基因在原核系统中的表达水平。(已经携带有氯霉素抗性质粒) 当要表达的蛋白质需要形成二硫键以形成正确的折叠时,可以选择K–12衍生菌Origami 2系列,thioredoxin reductase (trxB) 和glutathione